
Newborn screening
Encyclopedia
Newborn screening is the process by which infants are screened shortly after birth for a list of disorders that are treatable, but difficult or impossible to detect clinically. Screening programs are often run by state or national governing bodies with the goal of screening all infants born in the jurisdiction. Newborn screening originated when Robert Guthrie
developed a method to screen for phenylketonuria
, a disorder which could be managed by dietary adjustment if diagnosed early. Whole blood
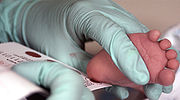
samples are collected from the infant's heel on specially designed filter paper
, and then tested for a panel of disorders. The disorders tested can vary from region to region, based on funding and the prevalence of a condition in the population.
is given much of the credit for pioneering the earliest screening for phenylketonuria
in the late 1960s using blood
samples on filter paper
obtained by pricking a newborn baby's heel on the second day of life to get a few drops of blood. Congenital hypothyroidism
was the second disease widely added in the 1970s. The development of tandem mass spectrometry
screening by Edwin Naylor and others in the early 1990s led to a large expansion of potentially detectable congenital metabolic diseases that affect blood levels of organic acids. Additional tests have been added to many screening programs. Confirmatory test for various metabolic disorders have evolved over the last two decade. First-line diagnosis in the organic acidemias is urine organic acid analysis by Gas chromatography-mass spectrometry (GC-MS), utilizing a capillary column. Organic acids can be measured in any physiologic fluid. However, it is most effective to use urine to identify the organic acids that signal these disorders, as semi-quantitative methods may not identify the important compounds in plasma. The organic acids found in the urine provide a high degree of suspicion for the specific pathway involved. The urinary organic acid profile is nearly always abnormal in the face of acute illness with decompensation. However, in some disorders the diagnostic analytes may be present only in small or barely detectable amounts when the affected individual is not acutely ill.
criteria used in newborn screening programs are based largely on criteria initially established by JMG Wilson and F. Jungner in 1968. Their publication, Principles and practice of screening for disease proposed ten criteria that screening programs should meet before being used as a public health
measure. The four criteria that are relied upon when making decisions for newborn screening programs are: having an acceptable treatment protocol in place that changes the outcome for patients diagnosed early with the disease, an understanding of the condition's natural history, an understanding about who will be treated as a patient, and a test that is reliable for both affected and unaffected patients and is acceptable to the public.
As diagnostic techniques have progressed, debates have arisen as to how screening programs should adapt. Tandem mass spectrometry
has greatly expanded the potential number of diseases that can be detected, even without satisfying all of the other criteria used for making screening decisions. Duchenne muscular dystrophy
is a disease that has been added to screening programs in several jurisdictions around the world, despite the lack of evidence as to whether early detection improves the clinical outcome for a patient.
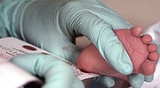
 Newborn screening tests are most commonly done from whole blood
Newborn screening tests are most commonly done from whole blood
samples collected on specially designed filter paper. The filter paper is often attached to a form containing required information about the infant and parents. This includes date and time of birth, date and time of sample collection, the infant's weight and gestational age. The form will also have information about whether the baby has had a blood transfusion and any additional nutrition the baby may have received (total parenteral nutrition
). Most newborn screening cards also include contact information for the infant's physician in cases where follow up screening or treatment is needed.
Ideally, newborn screening samples are collected from the infant between 24 hours and 7 days after birth. Samples can be collected at the hospital, or by midwives. If a sample is collected from an infant who is less than 24 hours old, the laboratory will often request a repeat specimen be taken after 24 hours. Samples are mailed daily to the laboratory responsible for testing. Most jurisdictions require samples to be collected for screening from all newborns, unless the parent or guardian opts out of the process in writing.
If an abnormality occurs, employees of the agency, usually nurses, begin to try to reach the physician, hospital, and/or nursery by telephone. They are persistent until they can arrange an evaluation of the infant by an appropriate specialist physician (depending on the disease). The specialist will attempt to confirm the diagnosis by repeating the tests by a different method or laboratory, or by performing other corroboratory or disproving tests. The confirmatory test varies depending on the initial screen, and can include enzyme assays, DNA testing, Gas Chromatography/Mass Spectrometry. Tandem mass spectrometry(MS/MS)is a screening step towards detection of the disorder. Depending on the likelihood of the diagnosis and the risk of delay, the specialist will initiate treatment and provide information to the family. Performance of the program is reviewed regularly and strenuous efforts are made to maintain a system that catches every infant with these diagnoses. Guidelines for newborn screening and follow up have been published by the American Academy of Pediatrics
.
The following conditions and disorders were recommended as "core panel" by the 2005 report of the American College of Medical Genetics (ACMG). The incidences reported below are from [ftp://ftp.hrsa.gov/mchb/genetics/screeningdraftforcomment.pdf their report], pages 143-307, though the rates may vary in different populations. (WARNING: The file is a very large PDF.)
Blood cell
disorders
Inborn errors of amino acid
metabolism
Inborn errors of organic acid
metabolism
Inborn errors of fatty acid metabolism
Miscellaneous multisystem diseases
Newborn screening by other methods than blood testing
Blood cell
disorders
Inborn errors of amino acid
metabolism
Inborn errors of organic acid
metabolism
Inborn errors of fatty acid metabolism
Congenital infections
Miscellaneous multisystem diseases
(MS/MS) equipment greatly increased the number of diseases that could be detected from the standard newborn screening blood spot card. MS/MS screening to determine concentrations of amino acid
s and acylcarnitines can be used to screen for a large number of inherited metabolic disorders.
Expanded newborn screening allowed a large number of disorders to be detected in a single test, previously newborn screening labs would have a single test for a single disease.
Instituting MS/MS screening often requires a sizable up front expenditure. When states choose to run their own programs the initial costs for equipment, training and new staff can be significant. Moreover, MS/MS gives only the screening result and not the confirmatory result. The same has to be further done by higher technologies or procedure like GC/MS, Enzyme Assays or DNA Tests. This in effect adds more cost burden and makes physicians lose precious time. To avoid at least a portion of the up front costs, some states such as Mississippi have chosen to contract with private labs for expanded screening. Others have chosen to form Regional Partnerships sharing both costs and resources. But for many states, screening is an integrated part of the department of health which can not or will not be easily replaced. Thus the initial expenditures can be difficult for states with tight budgets to justify. Screening fees have also increased in recent years as health care costs rise and more states add MS/MS screening to their programs. (See Report of Summation of Fees Charged for Newborn Screening, 2001–2005) Dollars spent for these programs may reduce resources available to other potentially lifesaving programs. It has been recommended that one disorder, Short Chain Acyl-coenzyme A Dehydrogenase Deficiency, or SCAD, be eliminated from screening programs, due to a "spurious association between SCAD and symptoms.
However, recent studies suggest that expanded screening is cost effective (see [ftp://ftp.hrsa.gov/mchb/genetics/screeningdraftforcomment.pdf ACMG report page 94-95] and articles published in Pediatrics. Advocates are quick to point out studies such as these when trying to convince state legislatures to mandate expanded screening.
Expanded newborn screening is also opposed by among some health care providers who are concerned that effective follow-up and treatment may not be available, that false positive screening tests may cause harm, and issues of informed consent
.
Controversy has also erupted in some countries over collection and storge of blood or DNA samples by government agencies during the routine newborn blood screen. It was revealed that in Texas the state had collected and stored blood and DNA samples on millions of newborns without the parents knowledge or consent. These samples were then used by the state for genetic experiments and to setup a database to catalog all of the samples/newborns. Samples obtained without parent's consent were destroyed.
Robert Guthrie
Dr. Robert Guthrie was an American microbiologist, the first to use dried blood spot testing, which he first did in the 1960's...
developed a method to screen for phenylketonuria
Phenylketonuria
Phenylketonuria is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase , rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine...
, a disorder which could be managed by dietary adjustment if diagnosed early. Whole blood
Whole blood
Whole blood is a term used in transfusion medicine for human blood from a standard blood donation. The blood is typically combined with an anticoagulant during the collection process, but is generally otherwise unprocessed...
samples are collected from the infant's heel on specially designed filter paper
Filter paper
Filter paper is a semi-permeable paper barrier placed perpendicular to a liquid or air flow. It is used to separate fine solids from liquids or air.-Properties:Filter paper comes in various porosities and grades depending on the applications it is meant for...
, and then tested for a panel of disorders. The disorders tested can vary from region to region, based on funding and the prevalence of a condition in the population.
Goals
Universal newborn screening (NBS) aims to identify infants that appear healthy at birth, but are afflicted with conditions that can cause severe illness or death. With early detection, these conditions can be managed to prevent complications.History
Robert GuthrieRobert Guthrie
Dr. Robert Guthrie was an American microbiologist, the first to use dried blood spot testing, which he first did in the 1960's...
is given much of the credit for pioneering the earliest screening for phenylketonuria
Phenylketonuria
Phenylketonuria is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase , rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine...
in the late 1960s using blood
Blood
Blood is a specialized bodily fluid in animals that delivers necessary substances such as nutrients and oxygen to the cells and transports metabolic waste products away from those same cells....
samples on filter paper
Filter paper
Filter paper is a semi-permeable paper barrier placed perpendicular to a liquid or air flow. It is used to separate fine solids from liquids or air.-Properties:Filter paper comes in various porosities and grades depending on the applications it is meant for...
obtained by pricking a newborn baby's heel on the second day of life to get a few drops of blood. Congenital hypothyroidism
Congenital hypothyroidism
Congenital hypothyroidism is a condition of thyroid hormone deficiency present at birth. Approximately 1 in 4000 newborn infants has a severe deficiency of thyroid function, while even more have mild or partial degrees. If untreated for several months after birth, severe congenital hypothyroidism...
was the second disease widely added in the 1970s. The development of tandem mass spectrometry
Tandem mass spectrometry
Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages.-Tandem MS instruments:...
screening by Edwin Naylor and others in the early 1990s led to a large expansion of potentially detectable congenital metabolic diseases that affect blood levels of organic acids. Additional tests have been added to many screening programs. Confirmatory test for various metabolic disorders have evolved over the last two decade. First-line diagnosis in the organic acidemias is urine organic acid analysis by Gas chromatography-mass spectrometry (GC-MS), utilizing a capillary column. Organic acids can be measured in any physiologic fluid. However, it is most effective to use urine to identify the organic acids that signal these disorders, as semi-quantitative methods may not identify the important compounds in plasma. The organic acids found in the urine provide a high degree of suspicion for the specific pathway involved. The urinary organic acid profile is nearly always abnormal in the face of acute illness with decompensation. However, in some disorders the diagnostic analytes may be present only in small or barely detectable amounts when the affected individual is not acutely ill.
Disease qualification
ScreeningScreening (medicine)
Screening, in medicine, is a strategy used in a population to detect a disease in individuals without signs or symptoms of that disease. Unlike what generally happens in medicine, screening tests are performed on persons without any clinical sign of disease....
criteria used in newborn screening programs are based largely on criteria initially established by JMG Wilson and F. Jungner in 1968. Their publication, Principles and practice of screening for disease proposed ten criteria that screening programs should meet before being used as a public health
Public health
Public health is "the science and art of preventing disease, prolonging life and promoting health through the organized efforts and informed choices of society, organizations, public and private, communities and individuals" . It is concerned with threats to health based on population health...
measure. The four criteria that are relied upon when making decisions for newborn screening programs are: having an acceptable treatment protocol in place that changes the outcome for patients diagnosed early with the disease, an understanding of the condition's natural history, an understanding about who will be treated as a patient, and a test that is reliable for both affected and unaffected patients and is acceptable to the public.
As diagnostic techniques have progressed, debates have arisen as to how screening programs should adapt. Tandem mass spectrometry
Tandem mass spectrometry
Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages.-Tandem MS instruments:...
has greatly expanded the potential number of diseases that can be detected, even without satisfying all of the other criteria used for making screening decisions. Duchenne muscular dystrophy
Duchenne muscular dystrophy
Duchenne muscular dystrophy is a recessive X-linked form of muscular dystrophy, which results in muscle degeneration, difficulty walking, breathing, and death. The incidence is 1 in 3,000 boys. Females and males are affected, though females are rarely affected and are more often carriers...
is a disease that has been added to screening programs in several jurisdictions around the world, despite the lack of evidence as to whether early detection improves the clinical outcome for a patient.
Sample collection
Whole blood
Whole blood is a term used in transfusion medicine for human blood from a standard blood donation. The blood is typically combined with an anticoagulant during the collection process, but is generally otherwise unprocessed...
samples collected on specially designed filter paper. The filter paper is often attached to a form containing required information about the infant and parents. This includes date and time of birth, date and time of sample collection, the infant's weight and gestational age. The form will also have information about whether the baby has had a blood transfusion and any additional nutrition the baby may have received (total parenteral nutrition
Total parenteral nutrition
Parenteral nutrition is feeding a person intravenously, bypassing the usual process of eating and digestion. The person receives nutritional formulae that contain nutrients such as glucose, amino acids, lipids and added vitamins and dietary minerals...
). Most newborn screening cards also include contact information for the infant's physician in cases where follow up screening or treatment is needed.
Ideally, newborn screening samples are collected from the infant between 24 hours and 7 days after birth. Samples can be collected at the hospital, or by midwives. If a sample is collected from an infant who is less than 24 hours old, the laboratory will often request a repeat specimen be taken after 24 hours. Samples are mailed daily to the laboratory responsible for testing. Most jurisdictions require samples to be collected for screening from all newborns, unless the parent or guardian opts out of the process in writing.
Reporting results
The goal is to report the results within a short period of time. If screens are normal, a paper report is sent to the submitting hospital and parents rarely hear about it.If an abnormality occurs, employees of the agency, usually nurses, begin to try to reach the physician, hospital, and/or nursery by telephone. They are persistent until they can arrange an evaluation of the infant by an appropriate specialist physician (depending on the disease). The specialist will attempt to confirm the diagnosis by repeating the tests by a different method or laboratory, or by performing other corroboratory or disproving tests. The confirmatory test varies depending on the initial screen, and can include enzyme assays, DNA testing, Gas Chromatography/Mass Spectrometry. Tandem mass spectrometry(MS/MS)is a screening step towards detection of the disorder. Depending on the likelihood of the diagnosis and the risk of delay, the specialist will initiate treatment and provide information to the family. Performance of the program is reviewed regularly and strenuous efforts are made to maintain a system that catches every infant with these diagnoses. Guidelines for newborn screening and follow up have been published by the American Academy of Pediatrics
American Academy of Pediatrics
The American Academy of Pediatrics is the major professional association of pediatricians in the United States. The AAP was founded in 1930 by 35 pediatricians to address pediatric healthcare standards. It currently has 60,000 members in primary care and sub-specialist areas...
.
Targeted disorders
The following list includes most of the disorders detected by the expanded or supplemental newborn screening by mass spectrometry. This expanded screening is not yet universally mandated by most states, but may be privately purchased by parents or hospitals at a cost of approximately US$80. The same can also be purchased from other countries like Germany, Austria, Spain , Japan and India where more than 100 disorders are being tested based on a urine sample of the newborn. Perhaps one in 5,000 infants will be positive for one of the metabolic tests below (excluding the congenital infections).Core panel
The following conditions and disorders were recommended as "core panel" by the 2005 report of the American College of Medical Genetics (ACMG). The incidences reported below are from [ftp://ftp.hrsa.gov/mchb/genetics/screeningdraftforcomment.pdf their report], pages 143-307, though the rates may vary in different populations. (WARNING: The file is a very large PDF.)
Blood cell
Blood cell
A blood cell, also called a hematocyte, is a cell normally found in blood. In mammals, these fall into three general categories:* red blood cells — Erythrocytes* white blood cells — Leukocytes* platelets — Thrombocytes...
disorders
- Sickle cell anemia (Hb SS) > 1 in 5,000; among African-Americans 1 in 400
- Sickle-cell diseaseSickle-cell diseaseSickle-cell disease , or sickle-cell anaemia or drepanocytosis, is an autosomal recessive genetic blood disorder with overdominance, characterized by red blood cells that assume an abnormal, rigid, sickle shape. Sickling decreases the cells' flexibility and results in a risk of various...
(Hb S/C) > 1 in 25,000 - Hb S/Beta-ThalassemiaThalassemiaThalassemia is an inherited autosomal recessive blood disease that originated in the Mediterranean region. In thalassemia the genetic defect, which could be either mutation or deletion, results in reduced rate of synthesis or no synthesis of one of the globin chains that make up hemoglobin...
(Hb S/Th) > 1 in 50,000
Inborn errors of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
metabolism
- Tyrosinemia I (TYR I) < 1 in 100,000
- Argininosuccinic aciduriaArgininosuccinic aciduriaArgininosuccinic aciduria, also called argininosuccinic acidemia, is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system...
(ASA) < 1 in 100,000 - CitrullinemiaCitrullinemiaCitrullinemia, also called citrullinuria, is an autosomal recessive urea cycle disorder that causes ammonia and other toxic substances to accumulate in the blood....
(CIT) < 1 in 100,000 - PhenylketonuriaPhenylketonuriaPhenylketonuria is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase , rendering it nonfunctional. This enzyme is necessary to metabolize the amino acid phenylalanine to the amino acid tyrosine...
(PKU) > 1 in 25,000 - Maple syrup urine diseaseMaple syrup urine diseaseMaple syrup urine disease , also called branched-chain ketoaciduria, is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia...
(MSUD) < 1 in 100,000 - HomocystinuriaHomocystinuriaHomocystinuria, also known as cystathionine beta synthase deficiency or CBS deficiency, is an inherited disorder of the metabolism of the amino acid methionine, often involving cystathionine beta synthase...
(HCY) < 1 in 100,000
Inborn errors of organic acid
Organic acid
An organic acid is an organic compound with acidic properties. The most common organic acids are the carboxylic acids, whose acidity is associated with their carboxyl group –COOH. Sulfonic acids, containing the group –SO2OH, are relatively stronger acids. The relative stability of the conjugate...
metabolism
- Glutaric acidemia type I (GA I) > 1 in 75,000
- Hydroxymethylglutaryl lyase deficiency (HMG) < 1 in 100,000
- Isovaleric acidemiaIsovaleric acidemiaIsovaleric acidemia, also called isovaleric aciduria or isovaleric acid CoA dehydrogenase deficiency, is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine...
(IVA) < 1 in 100,000 - 3-Methylcrotonyl-CoA carboxylase deficiency3-methylcrotonyl-CoA carboxylase deficiency3-Methylcrotonyl-CoA carboxylase deficiency , also known as 3-Methylcrotonylglycinuria or BMCC deficiency is an inherited disorder in which the body is unable to process certain proteins properly. People with this disorder have inadequate levels of an enzyme that helps break down proteins...
(3MCC) > 1 in 75,000 - Methylmalonyl-CoA mutase deficiencyMethylmalonyl-CoA mutase deficiencyMethylmalonyl-CoA mutase deficiency is an inborn error of organic acid metabolism. It is one of the 29 conditions currently recommended for newborn screening by the American College of Medical Genetics....
(MUT) > 1 in 75,000 - Methylmalonic aciduria, cblA and cblB forms (MMA, Cbl A,B) < 1 in 100,000
- Beta-ketothiolase deficiencyBeta-ketothiolase deficiencyBeta-ketothiolase deficiency is a rare, autosomal recessive metabolic disorder in which the body cannot properly process the amino acid isoleucine or the products of lipid breakdown....
(BKT) < 1 in 100,000 - Propionic acidemiaPropionic acidemiaPropionic acidemia, also known as propionic aciduria, propionyl-CoA carboxylase deficiency and ketotic glycinemia, is an autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia....
(PROP) > 1 in 75,000 - Multiple-CoA carboxylase deficiencyMultiple carboxylase deficiencyMultiple carboxylase deficiency is a form of metabolic disorder involving failures of carboxylation enzymes.The deficiency can be in biotinidase or holocarboxylase synthetase.These conditions respond to biotin.Forms include:...
(MCD) < 1 in 100,000
Inborn errors of fatty acid metabolism
- Long-chain hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) > 1 in 75,000
- Medium-chain acyl-CoA dehydrogenase deficiency (MCAD) > 1 in 25,000
- Very-long-chain acyl-CoA dehydrogenase deficiency (VLCAD) > 1 in 75,000
- Trifunctional protein deficiency (TFP) < 1 in 100,000
- Carnitine uptake defect (CUD) < 1 in 100,000
Miscellaneous multisystem diseases
- Cystic fibrosisCystic fibrosisCystic fibrosis is a recessive genetic disease affecting most critically the lungs, and also the pancreas, liver, and intestine...
(CF) > 1 in 5,000 - Congenital hypothyroidismCongenital hypothyroidismCongenital hypothyroidism is a condition of thyroid hormone deficiency present at birth. Approximately 1 in 4000 newborn infants has a severe deficiency of thyroid function, while even more have mild or partial degrees. If untreated for several months after birth, severe congenital hypothyroidism...
(CH) > 1 in 5,000 - Biotinidase deficiencyBiotinidase deficiencyBiotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency....
(BIOT) > 1 in 75,000 - Congenital adrenal hyperplasiaCongenital adrenal hyperplasiaCongenital adrenal hyperplasia refers to any of several autosomal recessive diseases resulting from mutations of genes for enzymes mediating the biochemical steps of production of cortisol from cholesterol by the adrenal glands ....
(CAH) > 1 in 25,000 - Classical galactosemia (GALT) > 1 in 50,000
Newborn screening by other methods than blood testing
- Congenital deafness (HEAR) > 1 in 5,000
Secondary targets
The following disorders are additional conditions that may be detected by screening. Many are listed as "secondary targets" by the 2005 report ACMG. Some states are now screening for more than 50 congenital conditions. Many of these are rare and unfamiliar to pediatricians and other primary health care professionals.Blood cell
Blood cell
A blood cell, also called a hematocyte, is a cell normally found in blood. In mammals, these fall into three general categories:* red blood cells — Erythrocytes* white blood cells — Leukocytes* platelets — Thrombocytes...
disorders
- Variant hemoglobinopathies (including Hb E)
- Glucose-6-phosphate dehydrogenase deficiencyGlucose-6-phosphate dehydrogenase deficiencyGlucose-6-phosphate dehydrogenase deficiency is an X-linked recessive hereditary disease characterised by abnormally low levels of glucose-6-phosphate dehydrogenase , a metabolic enzyme involved in the pentose phosphate pathway, especially important in red blood cell metabolism. G6PD deficiency is...
(G6PD)
Inborn errors of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
metabolism
- Tyrosinemia II
- ArgininemiaArgininemiaArgininemia, also called arginase deficiency, is an autosomal recessive urea cycle disorder where a deficiency of the enzyme arginase causes a build up of arginine and ammonia in the blood....
- Benign hyperphenylalaninemia
- Defects of biopterinBiopterinBiopterin is a coenzyme that is produced within the body.It is an oxidized degradation product of tetrahydrobiopterin.Defects in biopterin synthesis or regeneration can cause a form of hyperphenylalaninemia ....
cofactorCofactorCofactor may refer to any of the following:* Cofactor , the signed minor of a matrix* Minor , an alternative name for the determinant of a smaller matrix than that which it describes...
biosynthesis - Defects of biopterin cofactor regeneration
- Tyrosinemia III
- HypermethioninemiaHypermethioninemiaHypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.-Diagnosis:People with hypermethioninemia often do not show any symptoms...
- Citrullinemia type II
Inborn errors of organic acid
Organic acid
An organic acid is an organic compound with acidic properties. The most common organic acids are the carboxylic acids, whose acidity is associated with their carboxyl group –COOH. Sulfonic acids, containing the group –SO2OH, are relatively stronger acids. The relative stability of the conjugate...
metabolism
- Methylmalonic acidemiaMethylmalonic acidemiaMethylmalonic acidemia , also called methylmalonic aciduria, is an autosomal recessive metabolic disorder. It is a classical type of organic acidemia....
(Cbl C,D) - Malonic acidemia
- 2-Methyl 3-hydroxy butyric aciduria
- Isobutyryl-CoA dehydrogenase deficiency
- 2-Methylbutyryl-CoA dehydrogenase deficiency2-Methylbutyryl-CoA dehydrogenase deficiency2-Methylbutyryl-CoA dehydrogenase deficiency, also called 2-Methylbutyryl glycinuria or short/branched-chain acyl-CoA dehydrogenase deficiency , is an autosomal recessive metabolic disorder...
- 3-Methylglutaconyl-CoA hydratase deficiency3-methylglutaconic aciduria3-Methylglutaconic aciduria is used to describe at least five different disorders that impair the body's ability to make energy in the mitochondria. As a result of this impairment, 3-methylglutaconic acid and 3-methylglutaric acid build up and can be detected in the urine.3-Methylglutaconic acid...
- Glutaric acidemia type II
- HHH syndrome (Hyperammonemia, hyperornithinemia, homocitrullinuria syndrome)
- Beta-methyl crotonyl carboxylase deficiency
- Adenosylcobalamin synthesis defects
Inborn errors of fatty acid metabolism
- Medium/short-chain L-3-hydroxy acyl-CoA dehydrogenase deficiency
- Medium-chain ketoacyl-CoA thiolase deficiency
- Dienoyl-CoA reductase deficiency
- Glutaric acidemia type II
- Carnitine palmityl transferase deficiency type 1
- Carnitine palmityl transferase deficiency type 2
- Short-chain acyl-CoA dehydrogenase deficiency (SCAD)
- Carnitine/acylcarnitine Translocase Deficiency (Translocase)
- Short-chain hydroxy Acyl-CoA dehydrogenase deficiency (SCHAD)
- Long-chain acyl-CoA dehydrogenase deficiency (LCAD)
- Multiple acyl-CoA dehydrogenase deficiency (MADD)
Congenital infections
- TORCH complexTORCH complexTORCH complex is a medical acronym for a set of perinatal infections . The TORCH infections can lead to severe fetal anomalies or even fetal loss...
(ToxoplasmosisToxoplasmosisToxoplasmosis is a parasitic disease caused by the protozoan Toxoplasma gondii. The parasite infects most genera of warm-blooded animals, including humans, but the primary host is the felid family. Animals are infected by eating infected meat, by ingestion of feces of a cat that has itself...
, RubellaRubellaRubella, commonly known as German measles, is a disease caused by the rubella virus. The name "rubella" is derived from the Latin, meaning little red. Rubella is also known as German measles because the disease was first described by German physicians in the mid-eighteenth century. This disease is...
, CytomegalovirusCytomegalovirusCytomegalovirus is a viral genus of the viral group known as Herpesviridae or herpesviruses. It is typically abbreviated as CMV: The species that infects humans is commonly known as human CMV or human herpesvirus-5 , and is the most studied of all cytomegaloviruses...
, Herpes simplexHerpes simplexHerpes simplex is a viral disease caused by both Herpes simplex virus type 1 and type 2 . Infection with the herpes virus is categorized into one of several distinct disorders based on the site of infection. Oral herpes, the visible symptoms of which are colloquially called cold sores or fever...
, SyphilisSyphilisSyphilis is a sexually transmitted infection caused by the spirochete bacterium Treponema pallidum subspecies pallidum. The primary route of transmission is through sexual contact; however, it may also be transmitted from mother to fetus during pregnancy or at birth, resulting in congenital syphilis...
etc.), if there are indicative symptoms or mothers who may have been exposed - HIVHIVHuman immunodeficiency virus is a lentivirus that causes acquired immunodeficiency syndrome , a condition in humans in which progressive failure of the immune system allows life-threatening opportunistic infections and cancers to thrive...
Miscellaneous multisystem diseases
- Galactokinase deficiencyGalactokinase deficiencyGalactokinase deficiency, also known as Galactosemia type 2 or GALK deficiency, is an autosomal recessive metabolic disorder marked by an accumulation of galactose and galactitol secondary to the decreased conversion of galactose to galactose-1-phosphate by galactokinase...
- Galactose epimerase deficiencyGalactose epimerase deficiencyGalactose epimerase deficiency, also known as GALE deficiency, Galactosemia III and UDP-galactose-4-epimerase deficiency, is a rare, autosomal recessive form of galactosemia associated with a deficiency of the enzyme galactose epimerase....
- Maternal vitamin B12 deficiency
Expanded newborn screening
The increasing availability and decreasing cost of tandem mass spectrometryTandem mass spectrometry
Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages.-Tandem MS instruments:...
(MS/MS) equipment greatly increased the number of diseases that could be detected from the standard newborn screening blood spot card. MS/MS screening to determine concentrations of amino acid
Amino acid
Amino acids are molecules containing an amine group, a carboxylic acid group and a side-chain that varies between different amino acids. The key elements of an amino acid are carbon, hydrogen, oxygen, and nitrogen...
s and acylcarnitines can be used to screen for a large number of inherited metabolic disorders.
Expanded newborn screening allowed a large number of disorders to be detected in a single test, previously newborn screening labs would have a single test for a single disease.
Controversies
Newborn screening tests have become a subject of political controversy in the last decade. Two California babies, Zachary Wyvill and Zachary Black, were both born with Glutaric acidemia type I. Wyvill's birth hospital only tested for the four diseases mandated by state law, while Black was born at a hospital that was participating in an expanded testing pilot program. Black's disease was treated with diet and vitamins; Wyvill's disease went undetected for over six months, and during that time the damage from the enzyme deficiency became irreversible. Birth-defects lobbyists pushing for broader and more universal standards for newborn testing cite this as an example of how much of an impact testing can have.Instituting MS/MS screening often requires a sizable up front expenditure. When states choose to run their own programs the initial costs for equipment, training and new staff can be significant. Moreover, MS/MS gives only the screening result and not the confirmatory result. The same has to be further done by higher technologies or procedure like GC/MS, Enzyme Assays or DNA Tests. This in effect adds more cost burden and makes physicians lose precious time. To avoid at least a portion of the up front costs, some states such as Mississippi have chosen to contract with private labs for expanded screening. Others have chosen to form Regional Partnerships sharing both costs and resources. But for many states, screening is an integrated part of the department of health which can not or will not be easily replaced. Thus the initial expenditures can be difficult for states with tight budgets to justify. Screening fees have also increased in recent years as health care costs rise and more states add MS/MS screening to their programs. (See Report of Summation of Fees Charged for Newborn Screening, 2001–2005) Dollars spent for these programs may reduce resources available to other potentially lifesaving programs. It has been recommended that one disorder, Short Chain Acyl-coenzyme A Dehydrogenase Deficiency, or SCAD, be eliminated from screening programs, due to a "spurious association between SCAD and symptoms.
However, recent studies suggest that expanded screening is cost effective (see [ftp://ftp.hrsa.gov/mchb/genetics/screeningdraftforcomment.pdf ACMG report page 94-95] and articles published in Pediatrics. Advocates are quick to point out studies such as these when trying to convince state legislatures to mandate expanded screening.
Expanded newborn screening is also opposed by among some health care providers who are concerned that effective follow-up and treatment may not be available, that false positive screening tests may cause harm, and issues of informed consent
Informed consent
Informed consent is a phrase often used in law to indicate that the consent a person gives meets certain minimum standards. As a literal matter, in the absence of fraud, it is redundant. An informed consent can be said to have been given based upon a clear appreciation and understanding of the...
.
Controversy has also erupted in some countries over collection and storge of blood or DNA samples by government agencies during the routine newborn blood screen. It was revealed that in Texas the state had collected and stored blood and DNA samples on millions of newborns without the parents knowledge or consent. These samples were then used by the state for genetic experiments and to setup a database to catalog all of the samples/newborns. Samples obtained without parent's consent were destroyed.
External links
- U.S. National Newborn Screening and Genetics Resource Center
- The History of Newborn Screening - Flash Cast by Harvey Levy, MD: In this 40-minute talk and slide presentation, offered here in ten short video sections, Dr. Levy covers the history of newborn screening, starting with the origin of the concept of errors of inborn metabolism in the early 1900s, covering Dr. Robert Guthrie's development of newborn screening for PKU, and moving through current screening methods and public health approaches.
- Newborn Screening Information & Resources Homepage of the Save Babies Through Screening Foundation, a grass-roots advocacy group devoted solely to expanding, and promoting awareness of, Newborn Screening.
- About Newborn Screening
- Baily, M.A. and Murray, T.H. (2009).Ethics and Newborn Genetic Screening. Johns Hopkins University Press. ISBN 978-0-8018-9151-9
- PerkinElmer Genetics, Inc. (Commercial company that pioneered some of the screening procedures and offers testing directly to parents. Excellent set of links to other sites about metabolic diseases and screening.)
- Waldholz, Michael, "A Drop of Blood Saves One Baby; Another Falls Ill," Wall Street Journal, 17 June 2001, p. A1 (52k PDF)
- http://www.springerlink.com/content/024115x6215wj787/
- Novogenia DNA Plus Genetic Testing can save newborn
- Newborn Screening Program for Mumbai, Navi-Mumbai and Thane
- Baby's First Test (Educational site produced by the non-profit organization Genetic Alliance.)
- http://www.seefeldfuneral.com/obits/obituary.php?id=36042 Henry Morgan Anderson obituary
- http://www.adventuresofhenry.com Adventures of Henry, LLC publishing