
Mucopolysaccharidosis
Encyclopedia
Mucopolysaccharidoses are a group of metabolic disorders caused by the absence or malfunctioning of lysosomal
enzymes needed to break down molecules called glycosaminoglycans - long chains of sugar carbohydrates in each of our cells
that help build bone
, cartilage
, tendon
s, cornea
s, skin
and connective tissue
. Glycosaminoglycans (formerly called mucopolysaccharides) are also found in the fluid that lubricates our joint
s.
People with a mucopolysaccharidosis disease either do not produce enough of one of the 11 enzymes required to break down these sugar chains into simpler molecules, or they produce enzymes that do not work properly. Over time, these glycosaminoglycans collect in the cells, blood and connective tissues. The result is permanent, progressive cellular damage which affects appearance, physical abilities, organ and system functioning, and, in most cases, mental development.
The mucopolysaccharidoses are part of the lysosomal storage disease
family, a group of more than 40 genetic disorders that result when a specific organelle in our body's cells – the lysosome – malfunctions. The lysosome is commonly referred to as the cell’s recycling center because it processes unwanted material into substances that the cell can utilize. Lysosomes break down this unwanted matter via enzymes, highly specialized proteins essential for survival. Lysosomal disorders like mucopolysaccharidosis are triggered when a particular enzyme exists in too small an amount or is missing altogether.
The global mucopolysaccharidoses therapeutics market is attractive, with high unmet need. This unmet need in the market is 68%, valued at $0.44 billion according to Globaldata research analysis as of June 2011.
s (which send and receive signals throughout the body) as well as pain
and impaired motor function. This results from compression of nerves or nerve roots in the spinal cord
or in the peripheral nervous system
, the part of the nervous system
that connects the brain
and spinal cord
to sensory organs such as the eyes and to other organs, muscles, and tissues throughout the body.
Depending on the mucopolysaccharidosis subtype, affected individuals may have normal intellect or have cognitive impairments, may experience developmental delay, or may have severe behavioral problems. Many individuals have hearing loss, either conductive (in which pressure behind the ear drum causes fluid from the lining of the middle ear to build up and eventually congeal), neurosensitive (in which tiny hair cells in the inner ear are damaged), or both. Communicating hydrocephalus — in which the normal reabsorption of cerebrospinal fluid is blocked and causes increased pressure inside the head — is common in some of the mucopolysaccharidoses. Surgically inserting a shunt
into the brain can drain fluid. The eye's cornea
often becomes cloudy from intracellular storage, and glaucoma
and degeneration of the retina
also may affect the patient's vision.
Physical symptoms generally include coarse or rough facial features (including a flat nasal bridge, thick lips, and enlarged mouth and tongue), short stature with disproportionately short trunk (dwarfism
), dysplasia
(abnormal bone size and/or shape) and other skeletal irregularities, thickened skin, enlarged organs such as liver (hepatomegaly
) or spleen (splenomegaly
), hernias, and excessive body hair growth. Short and often claw-like hands, progressive joint stiffness, and carpal tunnel syndrome
can restrict hand mobility and function. Recurring respiratory infections are common, as are obstructive airway disease and obstructive sleep apnea
. Many affected individuals also have heart disease, often involving enlarged or diseased heart valves.
Another lysosomal storage disease often confused with the mucopolysaccharidoses is mucolipidosis
. In this disorder, excessive amounts of fatty materials known as lipids (another principal component of living cells) are stored, in addition to sugars. Persons with mucolipidosis may share some of the clinical features associated with the mucopolysaccharidoses (certain facial features, bony structure abnormalities, and damage to the brain), and increased amounts of the enzymes needed to break down the lipids are found in the blood.
alpha-L-iduronidase. Children born to an MPS I parent carry the defective gene
.
Affected children may be quite large at birth and appear normal but may have inguinal
(in the groin) or umbilical
(where the umbilical cord passes through the abdomen) hernias. Growth in height may be faster than normal but begins to slow before the end of the first year and often ends around age 3. Many children develop a short body trunk and a maximum stature of less than 4 feet. Distinct facial features (including flat face, depressed nasal bridge, and bulging forehead) become more evident in the second year. By age 2, the ribs have widened and are oar-shaped. The liver
, spleen
, and heart
are often enlarged. Children may experience noisy breathing and recurring upper respiratory tract and ear infections. Feeding may be difficult for some children, and many experience periodic bowel problems. Children with Hurler syndrome often die before age 10 from obstructive airway disease, respiratory infections, and cardiac complications.
Although no studies have been done to determine the frequency of MPS I in the United States, studies in British Columbia
estimate that 1 in 100,000 babies born has Hurler syndrome. The estimate for Scheie syndrome is one in 500,000 births and for Hurler-Scheie syndrome it is one in 115,000 births.
or iduronate sulfatase deficiency, is caused by lack of the enzyme iduronate sulfatase. Hunter syndrome has two clinical subtypes and (since it shows X-linked recessive inheritance) is the only one of the mucopolysaccharidoses in which the mother alone can pass the defective gene to a son. The incidence of Hunter syndrome is estimated to be 1 in 100,000 to 150,000 male births.
, is marked by severe neurological symptoms. These include progressive dementia
, aggressive behavior, hyperactivity, seizures, some deafness and loss of vision
, and an inability to sleep for more than a few hours at a time. This disorder tends to have three main stages. During the first stage, early mental and motor skill development may be somewhat delayed. Affected children show a marked decline in learning between ages 2 and 6, followed by eventual loss of language skills and loss of some or all hearing. Some children may never learn to speak. In the syndrome's second stage, aggressive behavior, hyperactivity, profound dementia, and irregular sleep may make children difficult to manage, particularly those who retain normal physical strength. In the syndrome's last stage, children become increasingly unsteady on their feet and most are unable to walk by age 10.
Thickened skin and mild changes in facial features, bone, and skeletal structures become noticeable with age. Growth in height usually stops by age 10. Other problems may include narrowing of the airway passage in the throat and enlargement of the tonsils and adenoids, making it difficult to eat or swallow. Recurring respiratory infections are common.
There are four distinct types of Sanfilippo syndrome, each caused by alteration of a different enzyme needed to completely break down the heparan sulfate
sugar chain. Little clinical difference exists between these four types but symptoms appear most severe and seem to progress more quickly in children with type A. The average duration of Sanfilippo syndrome is 8 to 10 years following onset of symptoms. Most persons with MPS III live into their teenage years, and some live longer.
The incidence of Sanfilippo syndrome (for all four types combined) is about one in 70,000 births.
, is estimated to occur in 1 in 700,000 births. Its two subtypes result from the missing or deficient enzymes galactose 6-sulfate sulfatase (Type A) or beta-galactosidase (Type B) needed to break down the keratan sulfate sugar chain. Clinical features are similar in both types but appear milder in Morquio Type B. Onset is between ages 1 and 3. Neurological complications include spinal nerve and nerve root compression resulting from extreme, progressive skeletal changes, particularly in the ribs and chest; conductive and/or neurosensitive loss of hearing and clouded corneas. Intelligence is normal unless hydrocephalus
develops and is not treated.
Physical growth slows and often stops between the ages of 4-8. Skeletal abnormalities include a bell-shaped chest, a flattening or curvature of the spine, shortened long bones, and dysplasia
of the hips, knees, ankles, and wrists. The bones that stabilize the connection between the head and neck can be malformed (odontoid hypoplasia); in these cases, a surgical procedure called spinal cervical bone fusion can be lifesaving. Restricted breathing, joint stiffness, and heart disease are also common. Children with the more severe form of Morquio syndrome may not live beyond their twenties or thirties.
. Caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase, Maroteaux-Lamy syndrome has a variable spectrum of severe symptoms. Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Growth is normal at first but stops suddenly around age 8. By age 10 children have developed a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction.
An enzyme replacement therapy was tested on patients with MPS VI and was successful in that it improved growth and joint movement. An experiment was then carried out to see whether an injection of the missing enzyme into the hips would help the range of motion and pain.
, one of the least common forms of the mucopolysaccharidoses, is estimated to occur in fewer than one in 250,000 births. The disorder is caused by deficiency of the enzyme beta-glucuronidase. In its rarest form, Sly syndrome causes children to be born with hydrops fetalis
, in which extreme amounts of fluid are retained in the body. Survival is usually a few months or less. Most children with Sly syndrome are less severely affected. Neurological symptoms may include mild to moderate mental retardation by age 3, communicating hydrocephalus, nerve entrapment, corneal clouding, and some loss of peripheral and night vision. Other symptoms include short stature, some skeletal irregularities, joint stiffness and restricted movement, and umbilical and/or inguinal hernias. Some patients may have repeated bouts of pneumonia during their first years of life. Most children with Sly syndrome live into the teenage or young adult years.
deficiency. Symptoms included nodular soft-tissue masses located around joints, with episodes of painful swelling of the masses and pain that ended spontaneously within 3 days. Pelvic radiography showed multiple soft-tissue masses and some bone erosion. Other traits included mild facial changes, acquired short stature as seen in other MPS disorders, and normal joint movement and intelligence.
and chorionic villus sampling
can verify if a fetus either carries a copy of the defective gene or is affected with the disorder. Genetic counseling can help parents who have a family history of the mucopolysaccharidoses determine if they are carrying the mutated gene that causes the disorders.
Changes to the diet will not prevent disease progression, but limiting milk, sugar, and dairy products has helped some individuals experiencing excessive mucus
.
Surgery to remove tonsils and adenoids may improve breathing among affected individuals with obstructive airway disorders and sleep apnea
. Sleep studies can assess airway status and the possible need for nighttime oxygen. Some patients may require surgical insertion of an endotrachial tube to aid breathing. Surgery can also correct hernias, help drain excessive cerebrospinal fluid from the brain, and free nerves and nerve roots compressed by skeletal and other abnormalities. Corneal transplants may improve vision among patients with significant corneal clouding.
Enzyme replacement therapy
(ERT) are currently in use or are being tested. Enzyme replacement therapy has proven useful in reducing non-neurological symptoms and pain. Currently BioMarin Pharmaceutical
produces enzyme replacement therapies for MPS type I and VI. In July 2006, the United States Food and Drug Administration
approved a synthetic version of I2S produced by Shire Pharmaceuticals Group
, called Elaprase, as a treatment for MPS type II (Hunter syndrome
).
Bone marrow transplantation (BMT) and umbilical cord blood transplantation (UCBT) have had limited success in treating the mucopolysaccharidoses. Abnormal physical characteristics, except for those affecting the skeleton and eyes, may be improved, but neurologic outcomes have varied. BMT and UCBT are high-risk procedures and are usually performed only after family members receive extensive evaluation and counseling.
For information on clinical trials
visit Clinical Trials Search
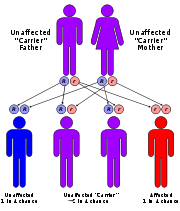 It is estimated that 1 in 25,000 babies born in the United States will have some form of the mucopolysaccharidoses. It is an autosomal recessive disorder, meaning that only individuals inheriting the defective gene from both parents are affected. (The exception is MPS II, or Hunter syndrome, in which the mother alone passes along the defective gene to a son.) When both people in a couple have the defective gene, each pregnancy carries with it a one in four chance that the child will be affected. The parents and siblings of an affected child may have no sign of the disorder. Unaffected siblings and select relatives of a child with one of the mucopolysaccharidoses may carry
It is estimated that 1 in 25,000 babies born in the United States will have some form of the mucopolysaccharidoses. It is an autosomal recessive disorder, meaning that only individuals inheriting the defective gene from both parents are affected. (The exception is MPS II, or Hunter syndrome, in which the mother alone passes along the defective gene to a son.) When both people in a couple have the defective gene, each pregnancy carries with it a one in four chance that the child will be affected. The parents and siblings of an affected child may have no sign of the disorder. Unaffected siblings and select relatives of a child with one of the mucopolysaccharidoses may carry
Lysosome
thumb|350px|Schematic of typical animal cell, showing subcellular components. [[Organelle]]s: [[nucleoli]] [[cell nucleus|nucleus]] [[ribosomes]] [[vesicle |vesicle]] rough [[endoplasmic reticulum]]...
enzymes needed to break down molecules called glycosaminoglycans - long chains of sugar carbohydrates in each of our cells
Cell (biology)
The cell is the basic structural and functional unit of all known living organisms. It is the smallest unit of life that is classified as a living thing, and is often called the building block of life. The Alberts text discusses how the "cellular building blocks" move to shape developing embryos....
that help build bone
Bone
Bones are rigid organs that constitute part of the endoskeleton of vertebrates. They support, and protect the various organs of the body, produce red and white blood cells and store minerals. Bone tissue is a type of dense connective tissue...
, cartilage
Cartilage
Cartilage is a flexible connective tissue found in many areas in the bodies of humans and other animals, including the joints between bones, the rib cage, the ear, the nose, the elbow, the knee, the ankle, the bronchial tubes and the intervertebral discs...
, tendon
Tendon
A tendon is a tough band of fibrous connective tissue that usually connects muscle to bone and is capable of withstanding tension. Tendons are similar to ligaments and fasciae as they are all made of collagen except that ligaments join one bone to another bone, and fasciae connect muscles to other...
s, cornea
Cornea
The cornea is the transparent front part of the eye that covers the iris, pupil, and anterior chamber. Together with the lens, the cornea refracts light, with the cornea accounting for approximately two-thirds of the eye's total optical power. In humans, the refractive power of the cornea is...
s, skin
Skin
-Dermis:The dermis is the layer of skin beneath the epidermis that consists of connective tissue and cushions the body from stress and strain. The dermis is tightly connected to the epidermis by a basement membrane. It also harbors many Mechanoreceptors that provide the sense of touch and heat...
and connective tissue
Connective tissue
"Connective tissue" is a fibrous tissue. It is one of the four traditional classes of tissues . Connective Tissue is found throughout the body.In fact the whole framework of the skeleton and the different specialized connective tissues from the crown of the head to the toes determine the form of...
. Glycosaminoglycans (formerly called mucopolysaccharides) are also found in the fluid that lubricates our joint
Joint
A joint is the location at which two or more bones make contact. They are constructed to allow movement and provide mechanical support, and are classified structurally and functionally.-Classification:...
s.
People with a mucopolysaccharidosis disease either do not produce enough of one of the 11 enzymes required to break down these sugar chains into simpler molecules, or they produce enzymes that do not work properly. Over time, these glycosaminoglycans collect in the cells, blood and connective tissues. The result is permanent, progressive cellular damage which affects appearance, physical abilities, organ and system functioning, and, in most cases, mental development.
The mucopolysaccharidoses are part of the lysosomal storage disease
Lysosomal storage disease
Lysosomal storage diseases are a group of approximately 50 rare inherited metabolic disorders that result from defects in lysosomal function...
family, a group of more than 40 genetic disorders that result when a specific organelle in our body's cells – the lysosome – malfunctions. The lysosome is commonly referred to as the cell’s recycling center because it processes unwanted material into substances that the cell can utilize. Lysosomes break down this unwanted matter via enzymes, highly specialized proteins essential for survival. Lysosomal disorders like mucopolysaccharidosis are triggered when a particular enzyme exists in too small an amount or is missing altogether.
The global mucopolysaccharidoses therapeutics market is attractive, with high unmet need. This unmet need in the market is 68%, valued at $0.44 billion according to Globaldata research analysis as of June 2011.
Features
The mucopolysaccharidoses share many clinical features but have varying degrees of severity. These features may not be apparent at birth but progress as storage of glycosaminoglycans affects bone, skeletal structure, connective tissues, and organs. Neurological complications may include damage to neuronNeuron
A neuron is an electrically excitable cell that processes and transmits information by electrical and chemical signaling. Chemical signaling occurs via synapses, specialized connections with other cells. Neurons connect to each other to form networks. Neurons are the core components of the nervous...
s (which send and receive signals throughout the body) as well as pain
Pain
Pain is an unpleasant sensation often caused by intense or damaging stimuli such as stubbing a toe, burning a finger, putting iodine on a cut, and bumping the "funny bone."...
and impaired motor function. This results from compression of nerves or nerve roots in the spinal cord
Spinal cord
The spinal cord is a long, thin, tubular bundle of nervous tissue and support cells that extends from the brain . The brain and spinal cord together make up the central nervous system...
or in the peripheral nervous system
Peripheral nervous system
The peripheral nervous system consists of the nerves and ganglia outside of the brain and spinal cord. The main function of the PNS is to connect the central nervous system to the limbs and organs. Unlike the CNS, the PNS is not protected by the bone of spine and skull, or by the blood–brain...
, the part of the nervous system
Nervous system
The nervous system is an organ system containing a network of specialized cells called neurons that coordinate the actions of an animal and transmit signals between different parts of its body. In most animals the nervous system consists of two parts, central and peripheral. The central nervous...
that connects the brain
Brain
The brain is the center of the nervous system in all vertebrate and most invertebrate animals—only a few primitive invertebrates such as sponges, jellyfish, sea squirts and starfishes do not have one. It is located in the head, usually close to primary sensory apparatus such as vision, hearing,...
and spinal cord
Spinal cord
The spinal cord is a long, thin, tubular bundle of nervous tissue and support cells that extends from the brain . The brain and spinal cord together make up the central nervous system...
to sensory organs such as the eyes and to other organs, muscles, and tissues throughout the body.
Depending on the mucopolysaccharidosis subtype, affected individuals may have normal intellect or have cognitive impairments, may experience developmental delay, or may have severe behavioral problems. Many individuals have hearing loss, either conductive (in which pressure behind the ear drum causes fluid from the lining of the middle ear to build up and eventually congeal), neurosensitive (in which tiny hair cells in the inner ear are damaged), or both. Communicating hydrocephalus — in which the normal reabsorption of cerebrospinal fluid is blocked and causes increased pressure inside the head — is common in some of the mucopolysaccharidoses. Surgically inserting a shunt
Shunt
Shunt may refer to:* Shunt - a hole or passage allowing fluid to move from one part of the body to another* Shunt - a device allowing electrical current to pass around a point in a circuit...
into the brain can drain fluid. The eye's cornea
Cornea
The cornea is the transparent front part of the eye that covers the iris, pupil, and anterior chamber. Together with the lens, the cornea refracts light, with the cornea accounting for approximately two-thirds of the eye's total optical power. In humans, the refractive power of the cornea is...
often becomes cloudy from intracellular storage, and glaucoma
Glaucoma
Glaucoma is an eye disorder in which the optic nerve suffers damage, permanently damaging vision in the affected eye and progressing to complete blindness if untreated. It is often, but not always, associated with increased pressure of the fluid in the eye...
and degeneration of the retina
Retina
The vertebrate retina is a light-sensitive tissue lining the inner surface of the eye. The optics of the eye create an image of the visual world on the retina, which serves much the same function as the film in a camera. Light striking the retina initiates a cascade of chemical and electrical...
also may affect the patient's vision.
Physical symptoms generally include coarse or rough facial features (including a flat nasal bridge, thick lips, and enlarged mouth and tongue), short stature with disproportionately short trunk (dwarfism
Dwarfism
Dwarfism is short stature resulting from a medical condition. It is sometimes defined as an adult height of less than 4 feet 10 inches , although this definition is problematic because short stature in itself is not a disorder....
), dysplasia
Dysplasia
Dysplasia , is a term used in pathology to refer to an abnormality of development. This generally consists of an expansion of immature cells, with a corresponding decrease in the number and location of mature cells. Dysplasia is often indicative of an early neoplastic process...
(abnormal bone size and/or shape) and other skeletal irregularities, thickened skin, enlarged organs such as liver (hepatomegaly
Hepatomegaly
Hepatomegaly is the condition of having an enlarged liver. It is a nonspecific medical sign having many causes, which can broadly be broken down into infection, direct toxicity, hepatic tumours, or metabolic disorder. Often, hepatomegaly will present as an abdominal mass...
) or spleen (splenomegaly
Splenomegaly
Splenomegaly is an enlargement of the spleen. The spleen usually lies in the left upper quadrant of the human abdomen. It is one of the four cardinal signs of hypersplenism, some reduction in the number of circulating blood cells affecting granulocytes, erythrocytes or platelets in any...
), hernias, and excessive body hair growth. Short and often claw-like hands, progressive joint stiffness, and carpal tunnel syndrome
Carpal tunnel syndrome
Carpal Tunnel Syndrome is an entrapment idiopathic median neuropathy, causing paresthesia, pain, and other symptoms in the distribution of the median nerve due to its compression at the wrist in the carpal tunnel. The pathophysiology is not completely understood but can be considered compression...
can restrict hand mobility and function. Recurring respiratory infections are common, as are obstructive airway disease and obstructive sleep apnea
Sleep apnea
Sleep apnea is a sleep disorder characterized by abnormal pauses in breathing or instances of abnormally low breathing, during sleep. Each pause in breathing, called an apnea, can last from a few seconds to minutes, and may occur 5 to 30 times or more an hour. Similarly, each abnormally low...
. Many affected individuals also have heart disease, often involving enlarged or diseased heart valves.
Another lysosomal storage disease often confused with the mucopolysaccharidoses is mucolipidosis
Mucolipidosis
Mucolipidosis is a group of inherited metabolic disorders that affect the body's ability to carry out the normal turnover of various materials within cells....
. In this disorder, excessive amounts of fatty materials known as lipids (another principal component of living cells) are stored, in addition to sugars. Persons with mucolipidosis may share some of the clinical features associated with the mucopolysaccharidoses (certain facial features, bony structure abnormalities, and damage to the brain), and increased amounts of the enzymes needed to break down the lipids are found in the blood.
Types
Seven distinct clinical types and numerous subtypes of the mucopolysaccharidoses have been identified. Although each mucopolysaccharidosis (MPS) differs clinically, most patients generally experience a period of normal development followed by a decline in physical and/or mental function. (Note: MPS-V and MPS-VIII are no longer in use as designations for any disease.)Overview table
| Type | Main diseases | Deficient enzyme | Accumulated products | Symptoms | Incidence |
|---|---|---|---|---|---|
| MPS I | Hurler syndrome Hurler syndrome Hurler syndrome, also known as mucopolysaccharidosis type I , Hurler's disease, also gargoylism, is a genetic disorder that results in the buildup of glycosaminoglycans due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of mucopolysaccharides in lysosomes... |
α-L-iduronidase |
|
Mental retardation Mental retardation is a generalized disorder appearing before adulthood, characterized by significantly impaired cognitive functioning and deficits in two or more adaptive behaviors... Macroglossia Macroglossia is the medical term for unusual enlargement of the tongue. Severe enlargement of the tongue can cause cosmetic and functional difficulties including in speaking, eating, swallowing and sleeping.- Amyloid Disorders :... Cardiomyopathy Cardiomyopathy, which literally means "heart muscle disease," is the deterioration of the function of the myocardium for any reason. People with cardiomyopathy are often at risk of arrhythmia or sudden cardiac death or both. Cardiomyopathy can often go undetected, making it especially dangerous to... Hepatosplenomegaly Hepatosplenomegaly is the simultaneous enlargement of both the liver and the spleen . Hepatosplenomegaly can occur as the result of acute viral hepatitis or infectious mononucleosis, or it can be the sign of a serious and life threatening lysosomal storage disease... |
1 in 100.000 |
| MPS II | Hunter syndrome Hunter syndrome Hunter syndrome, or mucopolysaccharidosis Type II, is a lysosomal storage disease caused by a deficient enzyme, iduronate-2-sulfatase . The syndrome is named after physician Charles A. Hunter , who first described it in 1917... |
Iduronate sulfatase |
Heparan sulfate Heparan sulfate is a linear polysaccharide found in all animal tissues. It occurs as a proteoglycan in which two or three HS chains are attached in close proximity to cell surface or extracellular matrix proteins... Dermatan sulfate Dermatan sulfate is a glycosaminoglycan found mostly in skin, but also in blood vessels, heart valves, tendons, and lungs.... |
Mental retardation Mental retardation is a generalized disorder appearing before adulthood, characterized by significantly impaired cognitive functioning and deficits in two or more adaptive behaviors... (similar, but milder, symptoms to Hurler syndrome. Is also X-linked recessive, as opposed to autosomal recessive.) |
~1 in 250.000 |
| MPS III | Sanfilippo syndrome Sanfilippo syndrome Sanfilippo syndrome, or Mucopolysaccharidosis III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate .Although undegraded heparan sulfate is the primary stored substrate,... A |
Heparan sulfamidase |
|
|
1 in 280,000 to 1 in 50,000 |
| Sanfilippo syndrome Sanfilippo syndrome Sanfilippo syndrome, or Mucopolysaccharidosis III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate .Although undegraded heparan sulfate is the primary stored substrate,... B |
N-acetylglucosaminidase | ||||
| Sanfilippo syndrome Sanfilippo syndrome Sanfilippo syndrome, or Mucopolysaccharidosis III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate .Although undegraded heparan sulfate is the primary stored substrate,... C |
Acetyl-CoA:alpha-glucosaminide acetyltransferase | ||||
| Sanfilippo syndrome Sanfilippo syndrome Sanfilippo syndrome, or Mucopolysaccharidosis III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate .Although undegraded heparan sulfate is the primary stored substrate,... D |
N-acetylglucosamine 6-sulfatase | ||||
| MPS IV | Morquio syndrome Morquio syndrome Morquio's syndrome is an autosomal recessive mucopolysaccharide storage disease , usually inherited. It is a rare type of birth defect with serious consequences... A |
Galactose-6-sulfate sulfatase |
Keratan sulfate Keratan sulfate , also called keratosulfate, is any of several sulfated glycosaminoglycans that have been found especially in the cornea, cartilage, and bone. It is also synthesized in the central nervous system where it participates both in development and in the glial scar formation following an... |
|
1 in 75,000 |
| Morquio syndrome Morquio syndrome Morquio's syndrome is an autosomal recessive mucopolysaccharide storage disease , usually inherited. It is a rare type of birth defect with serious consequences... B |
Beta-galactosidase Beta-galactosidase β-galactosidase, also called beta-gal or β-gal, is a hydrolase enzyme that catalyzes the hydrolysis of β-galactosides into monosaccharides. Substrates of different β-galactosidases include ganglioside GM1, lactosylceramides, lactose, and various glycoproteins... |
Keratan sulfate Keratan sulfate , also called keratosulfate, is any of several sulfated glycosaminoglycans that have been found especially in the cornea, cartilage, and bone. It is also synthesized in the central nervous system where it participates both in development and in the glial scar formation following an... |
|||
| MPS VI | Maroteaux-Lamy syndrome | N-acetylgalactosamine-4-sulfatase |
Dermatan sulfate Dermatan sulfate is a glycosaminoglycan found mostly in skin, but also in blood vessels, heart valves, tendons, and lungs.... |
Kyphosis Kyphosis , also called roundback or Kelso's hunchback, is a condition of over-curvature of the thoracic vertebrae... |
|
| MPS VII | Sly syndrome Sly syndrome Sly syndrome, also called Mucopolysaccharidosis Type VII or MPS, is an autosomal recessive lysosomal storage disease characterized by a deficiency of the enzyme β-glucuronidase, a lysosomal enzyme. Sly syndrome belongs to a group of disorders known as mucopolysaccharidoses, which are lysosomal... |
β-glucuronidase |
Heparan sulfate Heparan sulfate is a linear polysaccharide found in all animal tissues. It occurs as a proteoglycan in which two or three HS chains are attached in close proximity to cell surface or extracellular matrix proteins... Dermatan sulfate Dermatan sulfate is a glycosaminoglycan found mostly in skin, but also in blood vessels, heart valves, tendons, and lungs.... |
Hepatomegaly Hepatomegaly is the condition of having an enlarged liver. It is a nonspecific medical sign having many causes, which can broadly be broken down into infection, direct toxicity, hepatic tumours, or metabolic disorder. Often, hepatomegaly will present as an abdominal mass... |
Less than 1 in 250,000 |
| MPS IX | Natowicz syndrome | Hyaluronidase Hyaluronidase The hyaluronidases are a family of enzymes that degrade hyaluronic acid.In humans, there are six associated genes, including HYAL1, HYAL2, HYAL3, and PH-20/SPAM1.-Use as a drug:... |
|
|
|
MPS I
MPS I is divided into three subtypes based on severity of symptoms. All three types result from an absence of, or insufficient levels of, the enzymeEnzyme
Enzymes are proteins that catalyze chemical reactions. In enzymatic reactions, the molecules at the beginning of the process, called substrates, are converted into different molecules, called products. Almost all chemical reactions in a biological cell need enzymes in order to occur at rates...
alpha-L-iduronidase. Children born to an MPS I parent carry the defective gene
Gene
A gene is a molecular unit of heredity of a living organism. It is a name given to some stretches of DNA and RNA that code for a type of protein or for an RNA chain that has a function in the organism. Living beings depend on genes, as they specify all proteins and functional RNA chains...
.
- MPS I H (also called Hurler syndromeHurler syndromeHurler syndrome, also known as mucopolysaccharidosis type I , Hurler's disease, also gargoylism, is a genetic disorder that results in the buildup of glycosaminoglycans due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of mucopolysaccharides in lysosomes...
or α-L-iduronidase deficiency), is the most severe of the MPS I subtypes. Developmental delay is evident by the end of the first year, and patients usually stop developing between ages 2 and 4. This is followed by progressive mental decline and loss of physical skills. Language may be limited due to hearing loss and an enlarged tongue. In time, the clear layers of the cornea become clouded and retinas may begin to degenerate. Carpal tunnel syndrome (or similar compression of nerves elsewhere in the body) and restricted joint movement are common.
Affected children may be quite large at birth and appear normal but may have inguinal
Inguinal hernia
An inguinal hernia is a protrusion of abdominal-cavity contents through the inguinal canal. They are very common , and their repair is one of the most frequently performed surgical operations....
(in the groin) or umbilical
Umbilical hernia
Congenital umbilical hernia is a congenital malformation, common in infants of African descent. Among adults, it is three times more common in women than in men; among children, the ratio is roughly equal...
(where the umbilical cord passes through the abdomen) hernias. Growth in height may be faster than normal but begins to slow before the end of the first year and often ends around age 3. Many children develop a short body trunk and a maximum stature of less than 4 feet. Distinct facial features (including flat face, depressed nasal bridge, and bulging forehead) become more evident in the second year. By age 2, the ribs have widened and are oar-shaped. The liver
Liver
The liver is a vital organ present in vertebrates and some other animals. It has a wide range of functions, including detoxification, protein synthesis, and production of biochemicals necessary for digestion...
, spleen
Spleen
The spleen is an organ found in virtually all vertebrate animals with important roles in regard to red blood cells and the immune system. In humans, it is located in the left upper quadrant of the abdomen. It removes old red blood cells and holds a reserve of blood in case of hemorrhagic shock...
, and heart
Heart
The heart is a myogenic muscular organ found in all animals with a circulatory system , that is responsible for pumping blood throughout the blood vessels by repeated, rhythmic contractions...
are often enlarged. Children may experience noisy breathing and recurring upper respiratory tract and ear infections. Feeding may be difficult for some children, and many experience periodic bowel problems. Children with Hurler syndrome often die before age 10 from obstructive airway disease, respiratory infections, and cardiac complications.
- MPS I S, Scheie syndromeScheie syndromeScheie syndrome ") is less severe version of Hurler's disease. It is a condition characterized by corneal clouding, facial dysmorphism, and normal lifespan but unlike Hurler's, disease has normal intellect and ....
, is the mildest form of MPS I. Symptoms generally begin to appear after age 5, with diagnosis most commonly made after age 10. Children with Scheie syndrome have normal intelligence or may have mild learning disabilities; some may have psychiatric problems. GlaucomaGlaucomaGlaucoma is an eye disorder in which the optic nerve suffers damage, permanently damaging vision in the affected eye and progressing to complete blindness if untreated. It is often, but not always, associated with increased pressure of the fluid in the eye...
, retinal degeneration, and clouded corneas may significantly impair vision. Other problems include carpal tunnel syndrome or other nerve compression, stiff joints, claw hands and deformed feet, a short neck, and aortic valve disease. Some affected individuals also have obstructive airway disease and sleep apnea. Persons with Scheie syndrome can live into adulthood.
- MPS I H-S, Hurler-Scheie syndrome, is less severe than Hurler syndrome alone. Symptoms generally begin between ages 3 and 8. Children may have moderate mental retardation and learning difficulties. Skeletal and systemic irregularities include short stature, marked smallness in the jaws, progressive joint stiffness, compressed spinal cord, clouded corneas, hearing loss, heart disease, coarse facial features, and umbilical hernia. Respiratory problems, sleep apnea, and heart disease may develop in adolescence. Some persons with MPS I H-S need continuous positive airway pressure during sleep to ease breathing. Life expectancy is generally into the late teens or early twenties.
Although no studies have been done to determine the frequency of MPS I in the United States, studies in British Columbia
British Columbia
British Columbia is the westernmost of Canada's provinces and is known for its natural beauty, as reflected in its Latin motto, Splendor sine occasu . Its name was chosen by Queen Victoria in 1858...
estimate that 1 in 100,000 babies born has Hurler syndrome. The estimate for Scheie syndrome is one in 500,000 births and for Hurler-Scheie syndrome it is one in 115,000 births.
MPS II
MPS II, Hunter syndromeHunter syndrome
Hunter syndrome, or mucopolysaccharidosis Type II, is a lysosomal storage disease caused by a deficient enzyme, iduronate-2-sulfatase . The syndrome is named after physician Charles A. Hunter , who first described it in 1917...
or iduronate sulfatase deficiency, is caused by lack of the enzyme iduronate sulfatase. Hunter syndrome has two clinical subtypes and (since it shows X-linked recessive inheritance) is the only one of the mucopolysaccharidoses in which the mother alone can pass the defective gene to a son. The incidence of Hunter syndrome is estimated to be 1 in 100,000 to 150,000 male births.
MPS III
MPS III, Sanfilippo syndromeSanfilippo syndrome
Sanfilippo syndrome, or Mucopolysaccharidosis III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate .Although undegraded heparan sulfate is the primary stored substrate,...
, is marked by severe neurological symptoms. These include progressive dementia
Dementia
Dementia is a serious loss of cognitive ability in a previously unimpaired person, beyond what might be expected from normal aging...
, aggressive behavior, hyperactivity, seizures, some deafness and loss of vision
Visual perception
Visual perception is the ability to interpret information and surroundings from the effects of visible light reaching the eye. The resulting perception is also known as eyesight, sight, or vision...
, and an inability to sleep for more than a few hours at a time. This disorder tends to have three main stages. During the first stage, early mental and motor skill development may be somewhat delayed. Affected children show a marked decline in learning between ages 2 and 6, followed by eventual loss of language skills and loss of some or all hearing. Some children may never learn to speak. In the syndrome's second stage, aggressive behavior, hyperactivity, profound dementia, and irregular sleep may make children difficult to manage, particularly those who retain normal physical strength. In the syndrome's last stage, children become increasingly unsteady on their feet and most are unable to walk by age 10.
Thickened skin and mild changes in facial features, bone, and skeletal structures become noticeable with age. Growth in height usually stops by age 10. Other problems may include narrowing of the airway passage in the throat and enlargement of the tonsils and adenoids, making it difficult to eat or swallow. Recurring respiratory infections are common.
There are four distinct types of Sanfilippo syndrome, each caused by alteration of a different enzyme needed to completely break down the heparan sulfate
Heparan sulfate
Heparan sulfate is a linear polysaccharide found in all animal tissues. It occurs as a proteoglycan in which two or three HS chains are attached in close proximity to cell surface or extracellular matrix proteins...
sugar chain. Little clinical difference exists between these four types but symptoms appear most severe and seem to progress more quickly in children with type A. The average duration of Sanfilippo syndrome is 8 to 10 years following onset of symptoms. Most persons with MPS III live into their teenage years, and some live longer.
- Sanfilippo A is the most severe of the MPS III disorders and is caused by the missing or altered enzyme heparan N-sulfatase. Children with Sanfilippo A have the shortest survival rate among those with the MPS III disorders.
- Sanfilippo B is caused by the missing or deficient enzyme alpha-N-acetylglucosaminidase.
- Sanfilippo C results from the missing or altered enzyme acetyl-CoAlpha-glucosaminide acetyltransferase.
- Sanfilippo D is caused by the missing or deficient enzyme N-acetylglucosamine 6-sulfatase.
The incidence of Sanfilippo syndrome (for all four types combined) is about one in 70,000 births.
MPS IV
MPS IV, Morquio syndromeMorquio syndrome
Morquio's syndrome is an autosomal recessive mucopolysaccharide storage disease , usually inherited. It is a rare type of birth defect with serious consequences...
, is estimated to occur in 1 in 700,000 births. Its two subtypes result from the missing or deficient enzymes galactose 6-sulfate sulfatase (Type A) or beta-galactosidase (Type B) needed to break down the keratan sulfate sugar chain. Clinical features are similar in both types but appear milder in Morquio Type B. Onset is between ages 1 and 3. Neurological complications include spinal nerve and nerve root compression resulting from extreme, progressive skeletal changes, particularly in the ribs and chest; conductive and/or neurosensitive loss of hearing and clouded corneas. Intelligence is normal unless hydrocephalus
Hydrocephalus
Hydrocephalus , also known as "water in the brain," is a medical condition in which there is an abnormal accumulation of cerebrospinal fluid in the ventricles, or cavities, of the brain. This may cause increased intracranial pressure inside the skull and progressive enlargement of the head,...
develops and is not treated.
Physical growth slows and often stops between the ages of 4-8. Skeletal abnormalities include a bell-shaped chest, a flattening or curvature of the spine, shortened long bones, and dysplasia
Dysplasia
Dysplasia , is a term used in pathology to refer to an abnormality of development. This generally consists of an expansion of immature cells, with a corresponding decrease in the number and location of mature cells. Dysplasia is often indicative of an early neoplastic process...
of the hips, knees, ankles, and wrists. The bones that stabilize the connection between the head and neck can be malformed (odontoid hypoplasia); in these cases, a surgical procedure called spinal cervical bone fusion can be lifesaving. Restricted breathing, joint stiffness, and heart disease are also common. Children with the more severe form of Morquio syndrome may not live beyond their twenties or thirties.
MPS VI
Children with MPS VI, Maroteaux-Lamy syndrome, usually have normal intellectual development but share many of the physical symptoms found in Hurler syndromeHurler syndrome
Hurler syndrome, also known as mucopolysaccharidosis type I , Hurler's disease, also gargoylism, is a genetic disorder that results in the buildup of glycosaminoglycans due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of mucopolysaccharides in lysosomes...
. Caused by the deficient enzyme N-acetylgalactosamine 4-sulfatase, Maroteaux-Lamy syndrome has a variable spectrum of severe symptoms. Neurological complications include clouded corneas, deafness, thickening of the dura (the membrane that surrounds and protects the brain and spinal cord), and pain caused by compressed or traumatized nerves and nerve roots.
Growth is normal at first but stops suddenly around age 8. By age 10 children have developed a shortened trunk, crouched stance, and restricted joint movement. In more severe cases, children also develop a protruding abdomen and forward-curving spine. Skeletal changes (particularly in the pelvic region) are progressive and limit movement. Many children also have umbilical or inguinal hernias. Nearly all children have some form of heart disease, usually involving valve dysfunction.
An enzyme replacement therapy was tested on patients with MPS VI and was successful in that it improved growth and joint movement. An experiment was then carried out to see whether an injection of the missing enzyme into the hips would help the range of motion and pain.
MPS VII
MPS VII, Sly syndromeSly syndrome
Sly syndrome, also called Mucopolysaccharidosis Type VII or MPS, is an autosomal recessive lysosomal storage disease characterized by a deficiency of the enzyme β-glucuronidase, a lysosomal enzyme. Sly syndrome belongs to a group of disorders known as mucopolysaccharidoses, which are lysosomal...
, one of the least common forms of the mucopolysaccharidoses, is estimated to occur in fewer than one in 250,000 births. The disorder is caused by deficiency of the enzyme beta-glucuronidase. In its rarest form, Sly syndrome causes children to be born with hydrops fetalis
Hydrops fetalis
Hydrops fetalis is a condition in the fetus characterized by an accumulation of fluid, or edema, in at least two fetal compartments. By comparison, hydrops allantois or hydrops amnion are an accumulation of excessive fluid in the allantoic or amniotic space respectively.-Presentation:Locations can...
, in which extreme amounts of fluid are retained in the body. Survival is usually a few months or less. Most children with Sly syndrome are less severely affected. Neurological symptoms may include mild to moderate mental retardation by age 3, communicating hydrocephalus, nerve entrapment, corneal clouding, and some loss of peripheral and night vision. Other symptoms include short stature, some skeletal irregularities, joint stiffness and restricted movement, and umbilical and/or inguinal hernias. Some patients may have repeated bouts of pneumonia during their first years of life. Most children with Sly syndrome live into the teenage or young adult years.
MPS IX
As of 2001, only one case of MPS IX had been reported. The disorder results from hyaluronidaseHyaluronidase
The hyaluronidases are a family of enzymes that degrade hyaluronic acid.In humans, there are six associated genes, including HYAL1, HYAL2, HYAL3, and PH-20/SPAM1.-Use as a drug:...
deficiency. Symptoms included nodular soft-tissue masses located around joints, with episodes of painful swelling of the masses and pain that ended spontaneously within 3 days. Pelvic radiography showed multiple soft-tissue masses and some bone erosion. Other traits included mild facial changes, acquired short stature as seen in other MPS disorders, and normal joint movement and intelligence.
Diagnosis
Diagnosis often can be made through clinical examination and urine tests (excess mucopolysaccharides are excreted in the urine). Enzyme assays (testing a variety of cells or body fluids in culture for enzyme deficiency) are also used to provide definitive diagnosis of one of the mucopolysaccharidoses. Prenatal diagnosis using amniocentesisAmniocentesis
Amniocentesis is a medical procedure used in prenatal diagnosis of chromosomal abnormalities and fetal infections, in which a small amount of amniotic fluid, which contains fetal tissues, is sampled from the amnion or amniotic sac surrounding a developing fetus, and the fetal DNA is examined for...
and chorionic villus sampling
Chorionic villus sampling
Chorionic villus sampling , sometimes misspelled "chorionic villous sampling", is a form of prenatal diagnosis to determine chromosomal or genetic disorders in the fetus. It entails sampling of the chorionic villus and testing it...
can verify if a fetus either carries a copy of the defective gene or is affected with the disorder. Genetic counseling can help parents who have a family history of the mucopolysaccharidoses determine if they are carrying the mutated gene that causes the disorders.
Treatment
Currently there is no cure for these disorders. Medical care is directed at treating systemic conditions and improving the person's quality of life. Physical therapy and daily exercise may delay joint problems and improve the ability to move.Changes to the diet will not prevent disease progression, but limiting milk, sugar, and dairy products has helped some individuals experiencing excessive mucus
Mucus
In vertebrates, mucus is a slippery secretion produced by, and covering, mucous membranes. Mucous fluid is typically produced from mucous cells found in mucous glands. Mucous cells secrete products that are rich in glycoproteins and water. Mucous fluid may also originate from mixed glands, which...
.
Surgery to remove tonsils and adenoids may improve breathing among affected individuals with obstructive airway disorders and sleep apnea
Sleep apnea
Sleep apnea is a sleep disorder characterized by abnormal pauses in breathing or instances of abnormally low breathing, during sleep. Each pause in breathing, called an apnea, can last from a few seconds to minutes, and may occur 5 to 30 times or more an hour. Similarly, each abnormally low...
. Sleep studies can assess airway status and the possible need for nighttime oxygen. Some patients may require surgical insertion of an endotrachial tube to aid breathing. Surgery can also correct hernias, help drain excessive cerebrospinal fluid from the brain, and free nerves and nerve roots compressed by skeletal and other abnormalities. Corneal transplants may improve vision among patients with significant corneal clouding.
Enzyme replacement therapy
Enzyme replacement therapy
Enzyme replacement therapy is a medical treatment replacing an enzyme in patients in whom that particular enzyme is deficient or absent. Usually this is done by giving the patient an intravenous infusion containing the enzyme...
(ERT) are currently in use or are being tested. Enzyme replacement therapy has proven useful in reducing non-neurological symptoms and pain. Currently BioMarin Pharmaceutical
BioMarin Pharmaceutical
BioMarin Pharmaceutical Inc. is a biotechnology firm based in Novato, California. It has offices and facilities in the US, South America, Asia and Europe. BioMarin's core business and research is in enzyme replacement therapies...
produces enzyme replacement therapies for MPS type I and VI. In July 2006, the United States Food and Drug Administration
Food and Drug Administration
The Food and Drug Administration is an agency of the United States Department of Health and Human Services, one of the United States federal executive departments...
approved a synthetic version of I2S produced by Shire Pharmaceuticals Group
Shire Pharmaceuticals Group
Shire Plc is a Jersey-registered, Irish-headquartered manufacturer of pharmaceuticals. Originating in and with a large operational base in the United Kingdom, its brands and products include Adderall XR, Carbatrol, Fosrenol, and the recently released Vyvanse....
, called Elaprase, as a treatment for MPS type II (Hunter syndrome
Hunter syndrome
Hunter syndrome, or mucopolysaccharidosis Type II, is a lysosomal storage disease caused by a deficient enzyme, iduronate-2-sulfatase . The syndrome is named after physician Charles A. Hunter , who first described it in 1917...
).
Bone marrow transplantation (BMT) and umbilical cord blood transplantation (UCBT) have had limited success in treating the mucopolysaccharidoses. Abnormal physical characteristics, except for those affecting the skeleton and eyes, may be improved, but neurologic outcomes have varied. BMT and UCBT are high-risk procedures and are usually performed only after family members receive extensive evaluation and counseling.
For information on clinical trials
ClinicalTrials.gov
ClinicalTrials.gov is a registry of clinical trials. It is run by the United States National Library of Medicine at the National Institutes of Health, and is the largest clinical trials database, currently holding registrations from over 93,000 trials from more than 170 countries in the...
visit Clinical Trials Search
Genetics
External links
- Society for Mucopolysaccharide Diseases in the UK
- The National MPS Society in the US, combating MPS and ML (mucolipidosisMucolipidosisMucolipidosis is a group of inherited metabolic disorders that affect the body's ability to carry out the normal turnover of various materials within cells....
) - The Canadian Society for Mucopolysaccharide and Related Diseases
- Hong Kong Mucopolysaccharidoses & Rare Genetic Diseases Mutual Aid Group
- BioMarin Pharmaceuticals Publicly traded company involved in providing enzyme treatment for MPS.
- MPS Australia The Australian MPS Society is a non profit organisation formed by parents, relatives and friends of those suffering from MPS.
- Hide and Seek Foundation For Lysosomal Disease Research
- Clinical Trials Search