

OPLS
Encyclopedia
The OPLS force field
was developed by Prof. William L. Jorgensen
at Purdue University
and later at Yale University
.
:


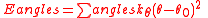
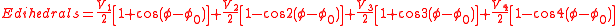

with the combining rules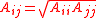 and
and 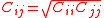 .
.
Intramolecular nonbonded interactions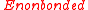 are counted only for atoms three or more bonds apart; 1,4 interactions are scaled down by the "fudge factor"
are counted only for atoms three or more bonds apart; 1,4 interactions are scaled down by the "fudge factor"  , otherwise
, otherwise  . All the interaction sites are centered on the atoms; there are no "lone pairs".
. All the interaction sites are centered on the atoms; there are no "lone pairs".
.
A distinctive feature of the OPLS parameters is that they were optimized to fit experimental properties of liquids, such as density and heat of vaporization, in addition to fitting gas-phase torsional profiles.
and MCPRO programs developed by Jorgensen. Other packages such as TINKER
, GROMACS, PCMODEL, Abalone
, Hyperchem, LAMMPS
and NAMD
also implement OPLS force fields.
Force field (chemistry)
In the context of molecular modeling, a force field refers to the form and parameters of mathematical functions used to describe the potential energy of a system of particles . Force field functions and parameter sets are derived from both experimental work and high-level quantum mechanical...
was developed by Prof. William L. Jorgensen
William L. Jorgensen
William L. Jorgensen is a Sterling Professor of Chemistry at Yale University. He is considered a pioneer in the field of computational chemistry...
at Purdue University
Purdue University
Purdue University, located in West Lafayette, Indiana, U.S., is the flagship university of the six-campus Purdue University system. Purdue was founded on May 6, 1869, as a land-grant university when the Indiana General Assembly, taking advantage of the Morrill Act, accepted a donation of land and...
and later at Yale University
Yale University
Yale University is a private, Ivy League university located in New Haven, Connecticut, United States. Founded in 1701 in the Colony of Connecticut, the university is the third-oldest institution of higher education in the United States...
.
Functional form
The functional form of the OPLS force field is very similar to that of AMBERAMBER
AMBER is a family of force fields for molecular dynamics of biomolecules originally developed by the late Peter Kollman's group at the University of California, San Francisco. AMBER is also the name for the molecular dynamics software package that simulates these force fields...
:
with the combining rules
and .Intramolecular nonbonded interactions
are counted only for atoms three or more bonds apart; 1,4 interactions are scaled down by the "fudge factor" , otherwise . All the interaction sites are centered on the atoms; there are no "lone pairs".Parameterization
Several sets of OPLS parameters have been published. There is OPLS-ua (united atom), which includes hydrogen atoms next to carbon implicitly in the carbon parameters, and can be used to save simulation time. OPLS-aa (all atom) includes every atom explicitly. Later publications include parameters for other specific functional groups and types of molecules such as carbohydrates. OPLS simulations in aqueous solution typically use the TIP4P or TIP3P water modelWater model
In computational chemistry, classical water models are used for the simulation of water clusters, liquid water, and aqueous solutions with explicit solvent. These models use the approximations of molecular mechanics...
.
A distinctive feature of the OPLS parameters is that they were optimized to fit experimental properties of liquids, such as density and heat of vaporization, in addition to fitting gas-phase torsional profiles.
Implementation
The reference implementations of the OPLS force field are the BOSSBOSS (molecular mechanics)
BOSS is a general-purpose molecular modeling program that performs molecular mechanics calculations, Metropolis Monte Carlo statistical mechanics simulations, and semiempirical AM1, PM3, and PDDG/PM3 quantum mechanics calculations...
and MCPRO programs developed by Jorgensen. Other packages such as TINKER
TINKER
TINKER is a computer software application for molecular dynamics simulation with a complete and general package for molecular mechanics and molecular dynamics, with some special features for biopolymers...
, GROMACS, PCMODEL, Abalone
Abalone (molecular mechanics)
Abalone is a general purpose molecular dynamics and molecular graphics program for simulations of bio-molecules in a periodic boundary conditions in explicit water or in implicit water models...
, Hyperchem, LAMMPS
LAMMPS
LAMMPS is a molecular dynamics program from Sandia National Laboratories.. LAMMPS makes use of MPI for parallel communication and is a free open-source code, distributed under the terms of the GNU General Public License.LAMMPS was originally developed under a Cooperative Research and Development...
and NAMD
NAMD
NAMD is a free-of-charge molecular dynamics simulation package written using the Charm++ parallel programming model, noted for its parallel efficiency and often used to simulate large systems...
also implement OPLS force fields.