

Shotgun sequencing
Encyclopedia
In genetics
, shotgun sequencing, also known as shotgun cloning, is a method used for sequencing
long DNA
strands. It is named by analogy with the rapidly-expanding, quasi-random firing pattern of a shotgun
.
Since the chain termination method of DNA sequencing
can only be used for fairly short strands (100 to 1000 basepairs), longer sequences must be subdivided into smaller fragments, and subsequently re-assembled to give the overall sequence. Two principal methods are used for this: chromosome walking, which progresses through the entire strand, piece by piece, and shotgun sequencing, which is a faster but more complex process, and uses random fragments.
In shotgun sequencing,
DNA is broken up randomly into numerous small segments, which are sequenced using the chain termination method to obtain reads. Multiple overlapping reads for the target DNA are obtained by performing several rounds of this fragmentation and sequencing. Computer programs then use the overlapping ends of different reads to assemble them into a continuous sequence.
Shotgun sequencing was one of the precursor technologies that was responsible for enabling full genome sequencing
.
In this extremely simplified example, none of the reads cover the full length of the original sequence, but the four reads can be assembled into the original sequence using the overlap of their ends to align and order them. In reality, this process uses enormous amounts of information that are rife with ambiguities and sequencing errors. Assembly of complex genomes is additionally complicated by the great abundance of repetitive sequence
, meaning similar short reads could come from completely different parts of the sequence.
Many overlapping reads for each segment of the original DNA are necessary to overcome these difficulties and accurately assemble the sequence. For example, to complete the Human Genome Project
, most of the human genome was sequenced at 12X or greater coverage; that is, each base in the final sequence was present, on average, in 12 reads. Even so, current methods have failed to isolate or assemble reliable sequence for approximately 1% of the (euchromatic
) human genome.
, known colloquially as double-barrel shotgun sequencing. As sequencing projects began to take on longer and more complicated DNAs, multiple groups began to realize that useful information could be obtained by sequencing both ends of a fragment of DNA. Although sequencing both ends of the same fragment and keeping track of the paired data was more cumbersome than sequencing a single end of two distinct fragments, the knowledge that the two sequences were oriented in opposite directions and were about the length of a fragment apart from each other was valuable in reconstructing the sequence of the original target fragment. The first published description of the use of paired ends was in 1990
as part of the sequencing of the human HGPRT
locus, although the use of paired ends was limited to closing gaps after the application of a traditional shotgun sequencing approach. The first theoretical description of a pure pairwise end sequencing strategy, assuming fragments of constant length, was in 1991. At the time, there was community consensus that the optimal fragment length for pairwise end sequencing would be three times the sequence read length. In 1995 Roach et al.
introduced the innovation of using fragments of varying sizes, and demonstrated that a pure pairwise end-sequencing strategy would be possible on large targets. The strategy was subsequently adopted by The Institute for Genomic Research
(TIGR) to sequence the genome of the bacterium Haemophilus influenzae
in 1995
, and then by Celera Genomics
to sequence the Drosophila melanogaster
(fruit fly) genome in 2000,
and subsequently the human genome.
To apply the strategy, high-molecular-weight DNA is sheared into random fragments, size-selected (usually 2, 10, 50, and 150 kb), and cloned into an appropriate vector. The clones are then sequenced from both ends using the chain termination method yielding two short sequences. Each sequence is called an end-read or read and two reads from the same clone are referred to as mate pairs
. Since the chain termination method usually can only produce reads between 500 and 1000 bases long, in all but the smallest clones, mate pairs
will rarely overlap.
The original sequence is reconstructed from the reads using sequence assembly software. First, overlapping reads are collected into longer composite sequences known as contig
s. Contigs can be linked together into scaffolds by following connections between mate pairs
. The distance between contigs can be inferred from the mate pair
positions if the average fragment length of the library is known and has a narrow window of deviation. Depending on the size of the gap between contigs, different techniques can be used to find the sequence in the gaps. If the gap is small (5-20kb) then the use of PCR to amplify the region is required, followed by sequencing. If the gap is large (>20kb) then the large fragment is cloned in special vectors such as BAC (Bacterial artificial chromosomes) followed by sequencing of the vector.
Proponents of this approach argue that it is possible to sequence the whole genome
at once using large arrays of sequencers, which makes the whole process much more efficient than more traditional approaches. Detractors argue that although the technique quickly sequences large regions of DNA, its ability to correctly link these regions is suspect, particularly for genomes with repeating regions. As sequence assembly
programs become more sophisticated and computing power becomes cheaper, it may be possible to overcome this limitation.
in the reconstructed sequence. It can be calculated from the length of the original genome (G), the number of reads(N), and the average read length(L) as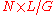 . For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy. This parameter also enables one to estimate other quantities, such as the percentage of the genome covered by reads (sometimes also called coverage). A high coverage in shotgun sequencing is desired because it can overcome errors in base calling and assembly. The subject of DNA sequencing theory
. For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy. This parameter also enables one to estimate other quantities, such as the percentage of the genome covered by reads (sometimes also called coverage). A high coverage in shotgun sequencing is desired because it can overcome errors in base calling and assembly. The subject of DNA sequencing theory
addresses the relationships of such quantities.
Sometimes a distinction is made between sequence coverage and physical coverage. Sequence coverage is the average number of times a base is read (as described above). Physical coverage is the average number of times a base is read or spanned by mate paired reads.
) was limited until the late 1990s, when technological advances made practical the handling of the vast quantities of complex data involved in the process. Historically, full-genome shotgun sequencing was believed to be limited by both the sheer size of large genomes and by the complexity added by the high percentage of repetitive DNA (greater than 50% for the human genome) present in large genomes. It was not widely accepted that a full-genome shotgun sequence of a large genome would provide reliable data. For these reasons, other strategies that lowered the computational load of sequence assembly had to be utilized before shotgun sequencing was performed.
In hierarchical sequencing, also known as top-down sequencing, a low-resolution physical map of the genome is made prior to actual sequencing. From this map, a minimal number of fragments that cover the entire chromosome are selected for sequencing. In this way, the minimum amount of high-throughput sequencing and assembly is required.
The amplified genome is first sheared into larger pieces (50-200kb) and cloned into a bacterial host using BACs
or PACs
. Because multiple genome copies have been sheared at random, the fragments contained in these clones have different ends, and with enough coverage (see section above) finding a scaffold of BAC contigs that covers the entire genome is theoretically possible. This scaffold is called a tiling path. Once a tiling path has been found, the BACs that form this path are sheared at random into smaller fragments and can be sequenced using the shotgun method on a smaller scale.
Although the full sequences of the BAC contigs is not known, their orientations relative to one another are known. There are several methods for deducing this order and selecting the BACs that make up a tiling path. The general strategy involves identifying the positions of the clones relative to one another and then selecting the least number of clones required to form a contiguous scaffold that covers the entire area of interest. The order of the clones is deduced by determining the way in which they overlap. Overlapping clones can be identified in several ways. A small radioactively- or chemically-labeled probe containing a sequence-tagged site
(STS) can be hybridized onto a microarray upon which the clones are printed. In this way, all the clones that contain a particular sequence in the genome are identified. The end of one of these clones can then be sequenced to yield a new probe and the process repeated in a method called chromosome walking. Alternatively, the BAC library can be restriction-digested. Two clones that have several fragment sizes in common are inferred to overlap because they contain multiple similarly spaced restriction sites in common. This method of genomic mapping is called restriction fingerprinting because it identifies a set of restriction sites contained in each clone. Once the overlap between the clones has found and their order relative to the genome known, a scaffold of a minimal subset of these contigs that covers the entire genome is shotgun-sequenced.
Because it involves first creating a low-resolution map of the genome, hierarchical shotgun sequencing is slower than whole-genome shotgun sequencing but relies less heavily on computer algorithms for genome assembly than whole-genome shotgun sequencing. The process of extensive BAC library creation and tiling path selection, however, make hierarchical shotgun sequencing slow and labor intensive. Now that the technology is available and the reliability of the data demonstrated, the speed and cost efficiency of whole-genome shotgun sequencing has made it the primary method for genome sequencing.
This results in high coverage, but the assembly process is much more computationally expensive. These technologies are vastly superior to shotgun sequencing due to the high volume of data and the relatively short time it takes to sequence a whole genome. The major disadvantage is that the accuracies are usually lower (although this is compensated for by the high coverage).
Genetics
Genetics , a discipline of biology, is the science of genes, heredity, and variation in living organisms....
, shotgun sequencing, also known as shotgun cloning, is a method used for sequencing
Sequencing
In genetics and biochemistry, sequencing means to determine the primary structure of an unbranched biopolymer...
long DNA
DNA
Deoxyribonucleic acid is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms . The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in...
strands. It is named by analogy with the rapidly-expanding, quasi-random firing pattern of a shotgun
Shotgun
A shotgun is a firearm that is usually designed to be fired from the shoulder, which uses the energy of a fixed shell to fire a number of small spherical pellets called shot, or a solid projectile called a slug...
.
Since the chain termination method of DNA sequencing
DNA sequencing
DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA....
can only be used for fairly short strands (100 to 1000 basepairs), longer sequences must be subdivided into smaller fragments, and subsequently re-assembled to give the overall sequence. Two principal methods are used for this: chromosome walking, which progresses through the entire strand, piece by piece, and shotgun sequencing, which is a faster but more complex process, and uses random fragments.
In shotgun sequencing,
DNA is broken up randomly into numerous small segments, which are sequenced using the chain termination method to obtain reads. Multiple overlapping reads for the target DNA are obtained by performing several rounds of this fragmentation and sequencing. Computer programs then use the overlapping ends of different reads to assemble them into a continuous sequence.
Shotgun sequencing was one of the precursor technologies that was responsible for enabling full genome sequencing
Full genome sequencing
Full genome sequencing , also known as whole genome sequencing , complete genome sequencing, or entire genome sequencing, is a laboratory process that determines the complete DNA sequence of an organism's genome at a single time...
.
Example
For example, consider the following two rounds of shotgun reads:| Strand | Sequence |
|---|---|
| Original | AGCATGCTGCAGTCATGCTTAGGCTA |
| First shotgun sequence | AGCATGCTGCAGTCATGCT------- |
| Second shotgun sequence | AGCATG-------------------- |
| Reconstruction | AGCATGCTGCAGTCATGCTTAGGCTA |
In this extremely simplified example, none of the reads cover the full length of the original sequence, but the four reads can be assembled into the original sequence using the overlap of their ends to align and order them. In reality, this process uses enormous amounts of information that are rife with ambiguities and sequencing errors. Assembly of complex genomes is additionally complicated by the great abundance of repetitive sequence
Repeated sequence (DNA)
In the study of DNA sequences, one can distinguish two main types of repeated sequence:*Tandem repeats:**Satellite DNA**Minisatellite**Microsatellite*Interspersed repeats:**SINEs...
, meaning similar short reads could come from completely different parts of the sequence.
Many overlapping reads for each segment of the original DNA are necessary to overcome these difficulties and accurately assemble the sequence. For example, to complete the Human Genome Project
Human Genome Project
The Human Genome Project is an international scientific research project with a primary goal of determining the sequence of chemical base pairs which make up DNA, and of identifying and mapping the approximately 20,000–25,000 genes of the human genome from both a physical and functional...
, most of the human genome was sequenced at 12X or greater coverage; that is, each base in the final sequence was present, on average, in 12 reads. Even so, current methods have failed to isolate or assemble reliable sequence for approximately 1% of the (euchromatic
Euchromatin
Euchromatin is a lightly packed form of chromatin that is rich in gene concentration, and is often under active transcription. Unlike heterochromatin, it is found in both cells with nuclei and cells without nuclei...
) human genome.
Whole genome shotgun sequencing
Whole genome shotgun sequencing for small (4000 to 7000 basepair) genomes was already in use in 1979. Broader application benefited from pairwise end sequencingPaired-end Tags
Paired-end tags, also known as PET, refer to the short sequences at the 5’ and 3’ ends of the DNA fragment of interest, which can be a piece of genomic DNA or cDNA. These short sequences are called tags or signatures because, in theory, they should contain enough sequence information to be uniquely...
, known colloquially as double-barrel shotgun sequencing. As sequencing projects began to take on longer and more complicated DNAs, multiple groups began to realize that useful information could be obtained by sequencing both ends of a fragment of DNA. Although sequencing both ends of the same fragment and keeping track of the paired data was more cumbersome than sequencing a single end of two distinct fragments, the knowledge that the two sequences were oriented in opposite directions and were about the length of a fragment apart from each other was valuable in reconstructing the sequence of the original target fragment. The first published description of the use of paired ends was in 1990
as part of the sequencing of the human HGPRT
Hypoxanthine-guanine phosphoribosyltransferase
Hypoxanthine-guanine phosphoribosyltransferase is an enzyme encoded in humans by the HPRT1 gene.HGPRT is a transferase that catalyzes conversion of hypoxanthine to inosine monophosphate and guanine to guanosine monophosphate. This reaction transfers the 5-phosphoribosyl group from...
locus, although the use of paired ends was limited to closing gaps after the application of a traditional shotgun sequencing approach. The first theoretical description of a pure pairwise end sequencing strategy, assuming fragments of constant length, was in 1991. At the time, there was community consensus that the optimal fragment length for pairwise end sequencing would be three times the sequence read length. In 1995 Roach et al.
introduced the innovation of using fragments of varying sizes, and demonstrated that a pure pairwise end-sequencing strategy would be possible on large targets. The strategy was subsequently adopted by The Institute for Genomic Research
The Institute for Genomic Research
The Institute for Genomic Research was a non-profit genomics research institute founded in 1992 by Craig Venter in Rockville, Maryland, United States. It is now a part of the J. Craig Venter Institute.-History:...
(TIGR) to sequence the genome of the bacterium Haemophilus influenzae
Haemophilus influenzae
Haemophilus influenzae, formerly called Pfeiffer's bacillus or Bacillus influenzae, Gram-negative, rod-shaped bacterium first described in 1892 by Richard Pfeiffer during an influenza pandemic. A member of the Pasteurellaceae family, it is generally aerobic, but can grow as a facultative anaerobe. H...
in 1995
, and then by Celera Genomics
Celera Genomics
Celera Corporation was a business unit of the Applera Corporation, but was spun off in July 2008 to become an independent publicly traded company. In May 2011 Quest Diagnostics Incorporated completed the acquisition of Celera, which thus became a wholly owned subsidiary...
to sequence the Drosophila melanogaster
Drosophila melanogaster
Drosophila melanogaster is a species of Diptera, or the order of flies, in the family Drosophilidae. The species is known generally as the common fruit fly or vinegar fly. Starting from Charles W...
(fruit fly) genome in 2000,
and subsequently the human genome.
To apply the strategy, high-molecular-weight DNA is sheared into random fragments, size-selected (usually 2, 10, 50, and 150 kb), and cloned into an appropriate vector. The clones are then sequenced from both ends using the chain termination method yielding two short sequences. Each sequence is called an end-read or read and two reads from the same clone are referred to as mate pairs
Paired-end Tags
Paired-end tags, also known as PET, refer to the short sequences at the 5’ and 3’ ends of the DNA fragment of interest, which can be a piece of genomic DNA or cDNA. These short sequences are called tags or signatures because, in theory, they should contain enough sequence information to be uniquely...
. Since the chain termination method usually can only produce reads between 500 and 1000 bases long, in all but the smallest clones, mate pairs
Paired-end Tags
Paired-end tags, also known as PET, refer to the short sequences at the 5’ and 3’ ends of the DNA fragment of interest, which can be a piece of genomic DNA or cDNA. These short sequences are called tags or signatures because, in theory, they should contain enough sequence information to be uniquely...
will rarely overlap.
The original sequence is reconstructed from the reads using sequence assembly software. First, overlapping reads are collected into longer composite sequences known as contig
Contig
A contig is a set of overlapping DNA segments that together represent a consensus region of DNA. In bottom-up sequencing projects, a contig refers to overlapping sequence data ; in top-down sequencing projects, contig refers to the overlapping clones that form a physical map of the genome that is...
s. Contigs can be linked together into scaffolds by following connections between mate pairs
Paired-end Tags
Paired-end tags, also known as PET, refer to the short sequences at the 5’ and 3’ ends of the DNA fragment of interest, which can be a piece of genomic DNA or cDNA. These short sequences are called tags or signatures because, in theory, they should contain enough sequence information to be uniquely...
. The distance between contigs can be inferred from the mate pair
Paired-end Tags
Paired-end tags, also known as PET, refer to the short sequences at the 5’ and 3’ ends of the DNA fragment of interest, which can be a piece of genomic DNA or cDNA. These short sequences are called tags or signatures because, in theory, they should contain enough sequence information to be uniquely...
positions if the average fragment length of the library is known and has a narrow window of deviation. Depending on the size of the gap between contigs, different techniques can be used to find the sequence in the gaps. If the gap is small (5-20kb) then the use of PCR to amplify the region is required, followed by sequencing. If the gap is large (>20kb) then the large fragment is cloned in special vectors such as BAC (Bacterial artificial chromosomes) followed by sequencing of the vector.
Proponents of this approach argue that it is possible to sequence the whole genome
Genome
In modern molecular biology and genetics, the genome is the entirety of an organism's hereditary information. It is encoded either in DNA or, for many types of virus, in RNA. The genome includes both the genes and the non-coding sequences of the DNA/RNA....
at once using large arrays of sequencers, which makes the whole process much more efficient than more traditional approaches. Detractors argue that although the technique quickly sequences large regions of DNA, its ability to correctly link these regions is suspect, particularly for genomes with repeating regions. As sequence assembly
Sequence assembly
In bioinformatics, sequence assembly refers to aligning and merging fragments of a much longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology cannot read whole genomes in one go, but rather reads small pieces of between 20 and 1000 bases,...
programs become more sophisticated and computing power becomes cheaper, it may be possible to overcome this limitation.
Coverage
Coverage is the average number of reads representing a given nucleotideNucleotide
Nucleotides are molecules that, when joined together, make up the structural units of RNA and DNA. In addition, nucleotides participate in cellular signaling , and are incorporated into important cofactors of enzymatic reactions...
in the reconstructed sequence. It can be calculated from the length of the original genome (G), the number of reads(N), and the average read length(L) as
. For example, a hypothetical genome with 2,000 base pairs reconstructed from 8 reads with an average length of 500 nucleotides will have 2x redundancy. This parameter also enables one to estimate other quantities, such as the percentage of the genome covered by reads (sometimes also called coverage). A high coverage in shotgun sequencing is desired because it can overcome errors in base calling and assembly. The subject of DNA sequencing theoryDNA sequencing theory
DNA sequencing theory is the broad body of work that attempts to lay analytical foundations for DNA sequencing. The practical aspects revolve around designing and optimizing sequencing projects , predicting project performance, troubleshooting experimental results, characterizing factors such as...
addresses the relationships of such quantities.
Sometimes a distinction is made between sequence coverage and physical coverage. Sequence coverage is the average number of times a base is read (as described above). Physical coverage is the average number of times a base is read or spanned by mate paired reads.
Hierarchical Shotgun sequencing
Although shotgun sequencing can in theory be applied to a genome of any size, its direct application to the sequencing of large genomes (for instance, the Human GenomeHuman genome
The human genome is the genome of Homo sapiens, which is stored on 23 chromosome pairs plus the small mitochondrial DNA. 22 of the 23 chromosomes are autosomal chromosome pairs, while the remaining pair is sex-determining...
) was limited until the late 1990s, when technological advances made practical the handling of the vast quantities of complex data involved in the process. Historically, full-genome shotgun sequencing was believed to be limited by both the sheer size of large genomes and by the complexity added by the high percentage of repetitive DNA (greater than 50% for the human genome) present in large genomes. It was not widely accepted that a full-genome shotgun sequence of a large genome would provide reliable data. For these reasons, other strategies that lowered the computational load of sequence assembly had to be utilized before shotgun sequencing was performed.
In hierarchical sequencing, also known as top-down sequencing, a low-resolution physical map of the genome is made prior to actual sequencing. From this map, a minimal number of fragments that cover the entire chromosome are selected for sequencing. In this way, the minimum amount of high-throughput sequencing and assembly is required.
The amplified genome is first sheared into larger pieces (50-200kb) and cloned into a bacterial host using BACs
Bacterial artificial chromosome
A bacterial artificial chromosome is a DNA construct, based on a functional fertility plasmid , used for transforming and cloning in bacteria, usually E. coli. F-plasmids play a crucial role because they contain partition genes that promote the even distribution of plasmids after bacterial cell...
or PACs
P1-derived artificial chromosome
The P1-derived artificial chromosome are DNA constructs that are derived from the DNA of P1 bacteriophage. They can carry large amounts of other sequences for a variety of bioengineering purposes...
. Because multiple genome copies have been sheared at random, the fragments contained in these clones have different ends, and with enough coverage (see section above) finding a scaffold of BAC contigs that covers the entire genome is theoretically possible. This scaffold is called a tiling path. Once a tiling path has been found, the BACs that form this path are sheared at random into smaller fragments and can be sequenced using the shotgun method on a smaller scale.
Although the full sequences of the BAC contigs is not known, their orientations relative to one another are known. There are several methods for deducing this order and selecting the BACs that make up a tiling path. The general strategy involves identifying the positions of the clones relative to one another and then selecting the least number of clones required to form a contiguous scaffold that covers the entire area of interest. The order of the clones is deduced by determining the way in which they overlap. Overlapping clones can be identified in several ways. A small radioactively- or chemically-labeled probe containing a sequence-tagged site
Sequence-tagged site
A sequence-tagged site is a short DNA sequence that has a single occurrence in the genome and whose location and base sequence are known....
(STS) can be hybridized onto a microarray upon which the clones are printed. In this way, all the clones that contain a particular sequence in the genome are identified. The end of one of these clones can then be sequenced to yield a new probe and the process repeated in a method called chromosome walking. Alternatively, the BAC library can be restriction-digested. Two clones that have several fragment sizes in common are inferred to overlap because they contain multiple similarly spaced restriction sites in common. This method of genomic mapping is called restriction fingerprinting because it identifies a set of restriction sites contained in each clone. Once the overlap between the clones has found and their order relative to the genome known, a scaffold of a minimal subset of these contigs that covers the entire genome is shotgun-sequenced.
Because it involves first creating a low-resolution map of the genome, hierarchical shotgun sequencing is slower than whole-genome shotgun sequencing but relies less heavily on computer algorithms for genome assembly than whole-genome shotgun sequencing. The process of extensive BAC library creation and tiling path selection, however, make hierarchical shotgun sequencing slow and labor intensive. Now that the technology is available and the reliability of the data demonstrated, the speed and cost efficiency of whole-genome shotgun sequencing has made it the primary method for genome sequencing.
Next-generation sequencing
Although shotgun sequencing was the most advanced technique for sequencing genomes from about 1995–2005, other technologies have surfaced, called next-generation sequencing. These technologies produce shorter reads (anywhere from 25–500bp) but many hundreds of thousands or millions of reads in a relatively short time (on the order of a day).This results in high coverage, but the assembly process is much more computationally expensive. These technologies are vastly superior to shotgun sequencing due to the high volume of data and the relatively short time it takes to sequence a whole genome. The major disadvantage is that the accuracies are usually lower (although this is compensated for by the high coverage).