

FreeON
Encyclopedia
FreeON is an experimental, open source (GPL) suite of programs for linear scaling quantum chemistry, formerly known as MondoSCF. It is highly modular, and has been written from scratch for N-scaling SCF theory in Fortran95
and C
. Platform independent IO is supported with HDF5. FreeON should compile with most modern Linux distributions. FreeON performs Hartree-Fock
, pure Density Functional
, and hybrid HF/DFT calculations (e.g. B3LYP) in a Cartesian-Gaussian LCAO basis. All algorithms are O(N) or O(N lg N) for non-metallic systems. Periodic boundary conditions in 1, 2 and 3 dimensions have been implemented through the Lorentz field (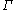 -point), and an internal coordinate geometry optimizer allows full (atom+cell) relaxation using analytic derivatives. Effective core potentials for energies and forces have been implemented, but Effective Core Potential (ECP) lattice forces do not work yet. Advanced features include O(N) static and dynamic response, as well as time reversible Born Oppenheimer Molecular Dynamics
-point), and an internal coordinate geometry optimizer allows full (atom+cell) relaxation using analytic derivatives. Effective core potentials for energies and forces have been implemented, but Effective Core Potential (ECP) lattice forces do not work yet. Advanced features include O(N) static and dynamic response, as well as time reversible Born Oppenheimer Molecular Dynamics
(MD).
Matt Challacombe, arXiv preprint 1001.2586
Fortran
Fortran is a general-purpose, procedural, imperative programming language that is especially suited to numeric computation and scientific computing...
and C
C (programming language)
C is a general-purpose computer programming language developed between 1969 and 1973 by Dennis Ritchie at the Bell Telephone Laboratories for use with the Unix operating system....
. Platform independent IO is supported with HDF5. FreeON should compile with most modern Linux distributions. FreeON performs Hartree-Fock
Hartree-Fock
In computational physics and chemistry, the Hartree–Fock method is an approximate method for the determination of the ground-state wave function and ground-state energy of a quantum many-body system....
, pure Density Functional
Density functional theory
Density functional theory is a quantum mechanical modelling method used in physics and chemistry to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. With this theory, the properties of a many-electron system can be determined by...
, and hybrid HF/DFT calculations (e.g. B3LYP) in a Cartesian-Gaussian LCAO basis. All algorithms are O(N) or O(N lg N) for non-metallic systems. Periodic boundary conditions in 1, 2 and 3 dimensions have been implemented through the Lorentz field (
-point), and an internal coordinate geometry optimizer allows full (atom+cell) relaxation using analytic derivatives. Effective core potentials for energies and forces have been implemented, but Effective Core Potential (ECP) lattice forces do not work yet. Advanced features include O(N) static and dynamic response, as well as time reversible Born Oppenheimer Molecular DynamicsMolecular dynamics
Molecular dynamics is a computer simulation of physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a period of time, giving a view of the motion of the atoms...
(MD).
Developers
| Matt Challacombe | Los Alamos National Laboratory |
| Eric Schwegler | Lawrence Livermore National Laboratory |
| C. J. Tymczak | Texas Southern University |
| Anders M. Niklasson | Los Alamos National Laboratory |
| Anders Odell | KTH Stockholm |
| Nicolas Bock | Los Alamos National Laboratory |
| Karoly Nemeth | Argonne National Laboratory |
| Valery Weber | University of Zurich |
| C. K. Gan | Institute for High Performance Computing |
| Graeme Henkelman | University of Texas at Austin |
Key Papers
- Linear Scaling Solution of the Time-Dependent Self-Consistent-Field Equations.
Matt Challacombe, arXiv preprint 1001.2586
- Representation independent algorithms for molecular response calculations in time-dependent self-consistent field theories. S. Tretiak, C. Isborn, AMN Niklasson and Matt Challacombe, Journal of Chemical Physics 130, 054111 (2009)
- Molecular orbital free algorithm for excited states in time-dependent perturbation theory. M. Lucero, AMN Niklasson, S. Tretiak and M. Challacombe, Journal of Chemical Physics, 129 064114 (2008)
- Time-reversible ab initio molecular dynamics A. Niklasson, C.J. Tymczak and Matt Challacombe, Journal of Chemical Physics, 126 144103 (2007)
- He conductivity in cool white dwarf atmospheres S. Mazevet, Matt Challacombe, P.M. Kowalski and D. Saumon Astrophysics and Space Science, 307 273 (2007)
- Time-reversible Born-Oppenheimer molecular dynamics A. Niklasson, C.J. Tymczak and Matt Challacombe, Physical Review Letters, 97 123001 (2006)
- Parallel algorithm for the computation of the Hartree-Fock exchange matrix: gas phase and periodic parallel ONX. V. Weber, Matt Challacombe, Journal of Chemical Physics, 125 104110 (2006)
- Energy gradients with respect to atomic positions and cell parameters for the Kohn-Sham density-functional theory at the Gamma point V. Weber, C.J. Tymczak and Matt Challacombe, Journal of Chemical Physics, 124 224107 (2006)
- Exchange energy gradients with respect to atomic positions and cell parameters within the Hartree-Fock {Gamma}-point approximation V. Weber, C. Daul and Matt Challacombe, Journal of Chemical Physics, 124 214105 (2006)
- Nonorthogonal density-matrix perturbation theory. A. Niklasson, V. Weber and Matt Challacombe, Journal of Chemical Physics, 123 44107 (2005)
- Higher-order response in O(N) by perturbed projection V. Weber, A. Niklasson, Matt Challacombe, Journal of Chemical Physics, 123 44106 (2005)
- Geometry optimization of crystals by the quasi-independent curvilinear coordinate approximation K. Nemeth and Matt Challacombe, Journal of Chemical Physics, 123 1 (2005)
- Linear scaling computation of the Fock matrix. VIII. Periodic boundaries for exact exchange at the Gamma point C.J. Tymczak, V. Weber, E. Schwegler and Matt Challacombe, Journal of Chemical Physics, 122 124105 (2005)
- Linear scaling computation of the Fock matrix. VII. Periodic density functional theory at the Gamma point C.J. Tymczak and Matt Challacombe, Journal of Chemical Physics, 122 134102 (2005)
- The quasi-independent curvilinear coordinate approximation for geometry optimization K. Nemeth and Matt Challacombe, Journal of Chemical Physics, 121 2877 (2004)
- Ab initio linear scaling response theory: Electric polarizability by perturbed projection V. Weber, A. Niklasson and Matt Challacombe, Physical Review Letters, 92 193002 (2004)
- Density matrix perturbation theory A. Niklasson and Matt Challacombe, Physical Review Letters, 92 193001 (2004)
- All-electron density-functional studies of hydrostatic compression of pentaerythritol tetranitrate C(CH2ONO2)4 C.K. Gan, T. Sewell and Matt Challacombe, Physical Review B, 69 35116 (2004)
- Linear scaling computation of Fock matrix. VII. Parallel computation of the Coulomb matrix, C. K. Gan, C. J. Tymczak, and Matt Challacombe, Journal of Chemical Physics, 121 (2004) 6608
- Linear scaling computation of the Fock matrix. VI. Data parallel computation of the exchange-correlation matrix C.K. Gan and Matt Challacombe, Journal of Chemical Physics, 118 9128 (2003)
- Trace resetting density matrix purification in O(N) self-consistent-field theory A. Niklasson, C.J. Tymczak and Matt Challacombe, Journal of Chemical Physics, 118 8611 (2003)
- Linear scaling computation of the Fock matrix. V. Hierarchical Cubature for numerical integration of the exchange-correlation matrix Matt Challacombe, Journal of Chemical Physics, 113 10037 (2000)
- Linear scaling computation of the Fock matrix. III. Formation of the exchange matrix with permutational symmetry E. Schwegler and Matt Challacombe, Theoretical Chemistry Accounts, 104 344 (2000)
- General parallel sparse-blocked matrix multiply for linear scaling SCF theory Matt Challacombe, Computer Physics Communications, 128 93 (2000)
- Linear scaling computation of the Fock matrix. IV. Multipole accelerated formation of the exchange matrix E. Schwegler and Matt Challacombe, Journal of Chemical Physics, 111 6223 (1999)
- A simplified density matrix minimization for linear scaling self-consistent field theory Matt Challacombe, Journal of Chemical Physics, 110 2332 (1999)
- A multipole acceptability criterion for electronic structure theory E. Schwegler, Matt Challacombe and M. Head-Gordon, Journal of Chemical Physics, 109 8764 (1998)
- Periodic boundary conditions and the fast multipole method Matt Challacombe, C. White and M. Head-Gordon, Journal of Chemical Physics, 107 10131 (1997)
- Linear scaling computation of the Fock matrix. II. Rigorous bounds on exchange integrals and incremental Fock build E. Schwegler, Matt Challacombe and M. Head-Gordon, Journal of Chemical Physics, 106 9708 (1997)
- Linear scaling computation of the Fock matrix Matt Challacombe and E. Schwegler, Journal of Chemical Physics, 106 5526 (1997)
- Linear scaling computation of the Hartree-Fock exchange matrix E. Schwegler and Matt Challacombe, Journal of Chemical Physics, 105 2726 (1996)
- Fast assembly of the Coulomb matrix: a quantum chemical tree code Matt Challacombe, E. Schwegler and J. Almlof, Journal of Chemical Physics, 104 4685 (1996)